Data analysis for Characterisation of the global transcriptional response to heat shock and the impact of individual genetic variation
Peter Humburg
Wellcome Trust Centre for Human Genetics, University of Oxford, Roosevelt Dr., Oxford, OX3 7BN, UKpeter.humburg@well.ox.ac.uk
Wanseon Lee
Wellcome Trust Centre for Human Genetics, University of Oxford, Roosevelt Dr., Oxford, OX3 7BN, UKwanseon.lee@well.ox.ac.uk
Sun 28 Aug 2016
Introduction
The heat shock response is a highly conserved mechanism responsible for the maintenance of cellular function under stress. Originally identified as part of the response to increased environmental temperatures (Ritossa 1962) heat shock proteins (HSPs) have since been implicated in susceptibility to infection (Kee et al. 2008), cancer risk (Sfar et al. 2010) and drug response (Martin et al. 2004). Despite its obvious significance many details of the heat shock response still remain uncertain.
Here we study the genome-wide changes to gene expression induced by heat shock in 60 HapMap cell lines from Yoruba individuals (Gibbs 2010) to identify genes and pathways involved in the human heat shock response. To further elucidate underlying mechanisms genetic variants modulating gene expression are mapped across all relevant genes.
Methods
Pre-processing and Quality Control
Raw gene expression QC
Probe intensities for resting and stimulated cells were imported into R for further processing together with associated meta data.
Annotations for all probes were obtained via the illuminaHumanv3.db Bioconductor package (Barbosa-Morais et al. 2010). Only probes that are considered to be of perfect or good quality were taken forward for analysis. Additionally, all probes mapping to more than one genomic location or to a location that contains a known SNP were excluded.
Probes were required to exhibit significant signal (detection p-value < 0.01) in at least 10 samples and samples with less than 30% of the remaining probes providing significant signal were excluded (together with the paired sample). Samples showing exceptionally low variation in probe intensities (standard deviation of the log intensities of all retained probes below 0.8) were also removed.
After filtering 12416 of 48803 probes (25.4%) and 56 samples remain. See Figure 1 for the distributions of intensities for the remaining probes in each sample.
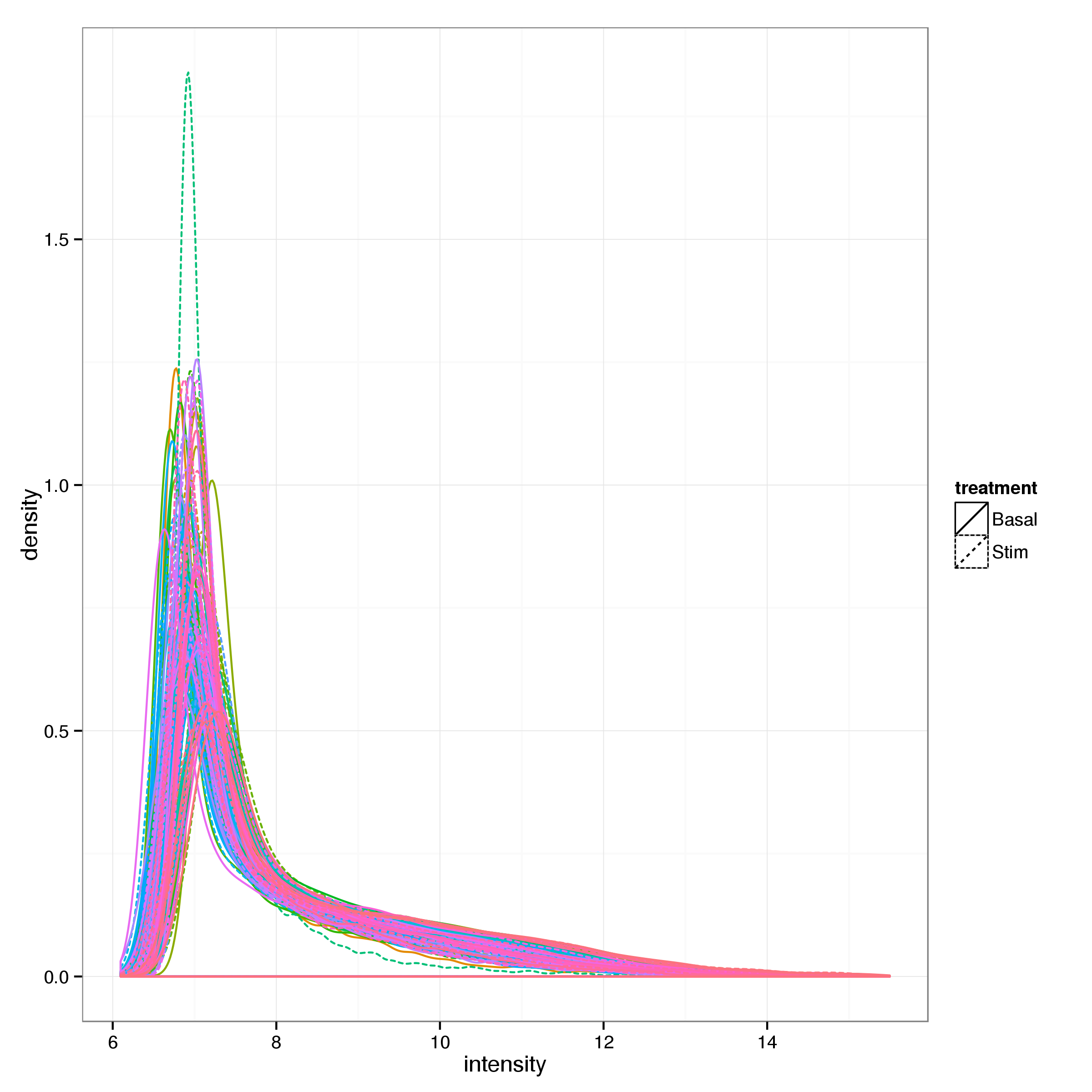
Figure 1: Distribution of probe intensities.
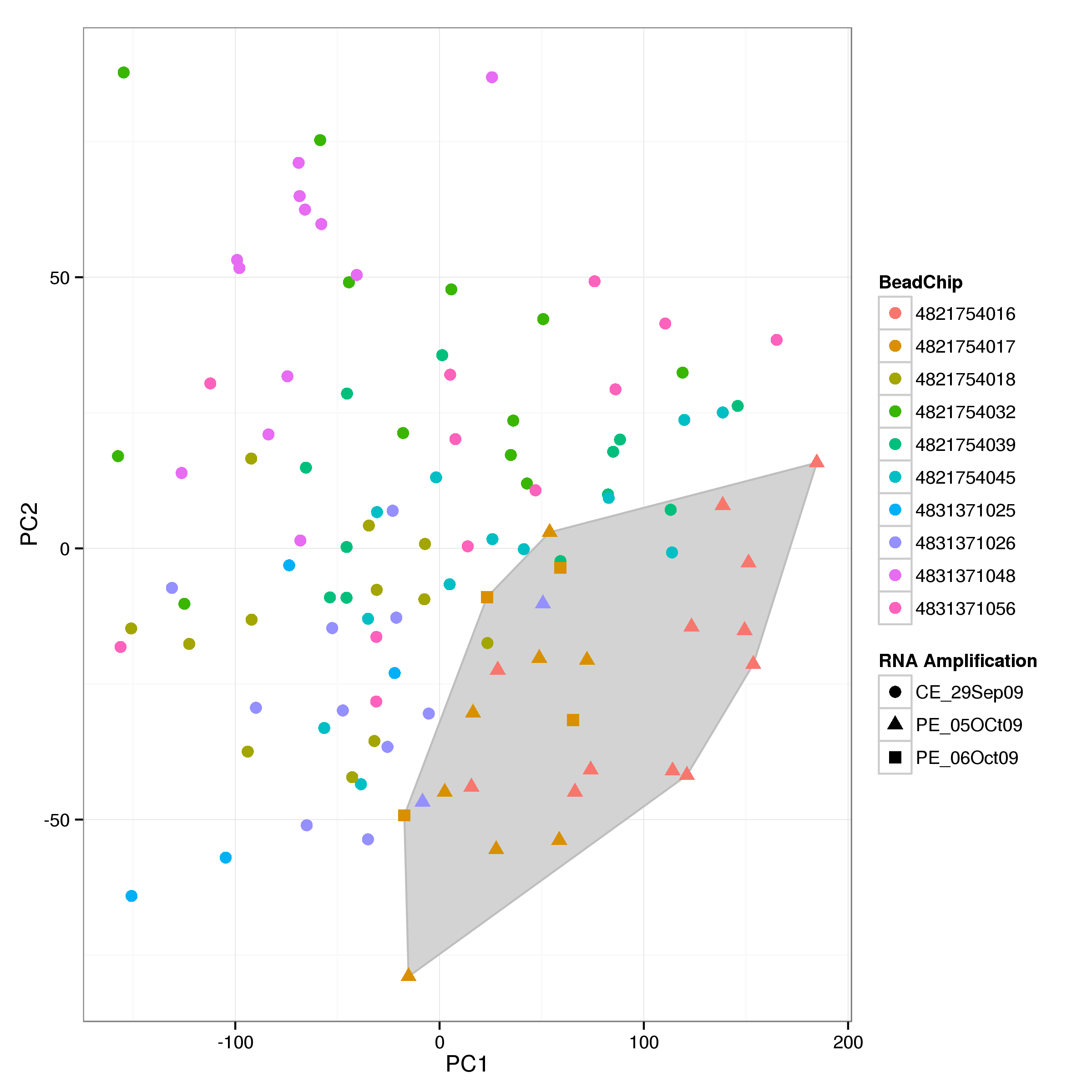
Figure 2: PCA plot of probe intensities by sample. Colours correspond to different BeadChips while shapes indicate different amplification dates. Samples from the two amplifications in October 2009 are clustered together (highlighted by the grey area).
A principle component analysis of the gene expression for the remaining samples indicates a clustering of samples from two RNA amplification batches in October 2009 (Figure 2). This batch effect is noted for later correction during the analysis.
Further analysis of the gene expression response to heat shock revealed that four of the samples (NA18502, NA18508, NA18522, NA18853, NA18504) exhibit patterns of expression inconsistent with those observed in other samples, including reversal of fold change for strongly differentially expressed genes. This suggests a potential mislabelling of these samples, which were consequently excluded.
Genotype QC
Genotype data provided by the HapMap project (Gibbs 2010) was processed with Plink (Chang et al. 2015) to restrict the data to autosomes and remove SNPs with low genotyping rate and and those with a minor allele frequency of less than 10% in the sample. This results in the exclusion of 794511 of 2582999 SNPs (30.76%);
The IBS nearest neighbour calculation shows that the majority of samples are quite similar with regard to the extend of genetic differences between them while a few show markedly increased sharing of alleles (see Figure 3 and Table 1). This is consistent with a population of largely unrelated individuals including a few individuals that are more closely related than expected. One individual (NA18913) also shows evidence of reduced similarity with the second and third nearest neighbour.
 for details). Note the spikes corresponding to individuals with positive Z-scores indicating increased similarity of their genomes compared to the rest of the cohort.](heatshock_analysis_files/figure-html/nnIBSplot-1.png)
Figure 3: Similarity between individuals. For each individual the similarity, in terms of IBS, to all other individuals is determined and individuals are ranked acording to this measure. From this a Z-score is computed for for each of the three closest neighbours (see the Plink documentation for details). Note the spikes corresponding to individuals with positive Z-scores indicating increased similarity of their genomes compared to the rest of the cohort.
| FID | IID | NN | MIN_DST | Z | FID2 | IID2 |
|---|---|---|---|---|---|---|
| Yoruba_028 | NA18913 | 1 | 0.8121 | 4.343 | Yoruba_117 | NA19238 |
| Yoruba_117 | NA19238 | 1 | 0.8121 | 4.343 | Yoruba_028 | NA18913 |
| Yoruba_024 | NA18862 | 3 | 0.7007 | 2.618 | Yoruba_048 | NA19203 |
| Yoruba_028 | NA18913 | 2 | 0.6985 | -2.340 | Yoruba_048 | NA19204 |
| Yoruba_028 | NA18913 | 3 | 0.6984 | -2.325 | Yoruba_112 | NA19192 |
| Yoruba_024 | NA18862 | 2 | 0.7009 | 2.183 | Yoruba_017 | NA18871 |
| Yoruba_101 | NA19130 | 1 | 0.7535 | 1.908 | Yoruba_112 | NA19192 |
| Yoruba_112 | NA19192 | 1 | 0.7535 | 1.908 | Yoruba_101 | NA19130 |
A similar picture emerges if we consider the estimated proportion of IBD for all sample pairs with 3 pairs showing evidence of higher than expected relatedness (see Table 2 and Figure 4 for details).
To ensure that at most one sample of each suspect pair are included in the analysis the samples NA18913, NA19192, NA18862 and NA19092 are excluded.
| IID1 | IID2 | PI_HAT | DST | RATIO |
|---|---|---|---|---|
| NA18913 | NA19238 | 0.5 | 0.812 | 76.8 |
| NA19092 | NA19101 | 0.104 | 0.723 | 2.82 |
| NA19130 | NA19192 | 0.239 | 0.753 | 4.5 |
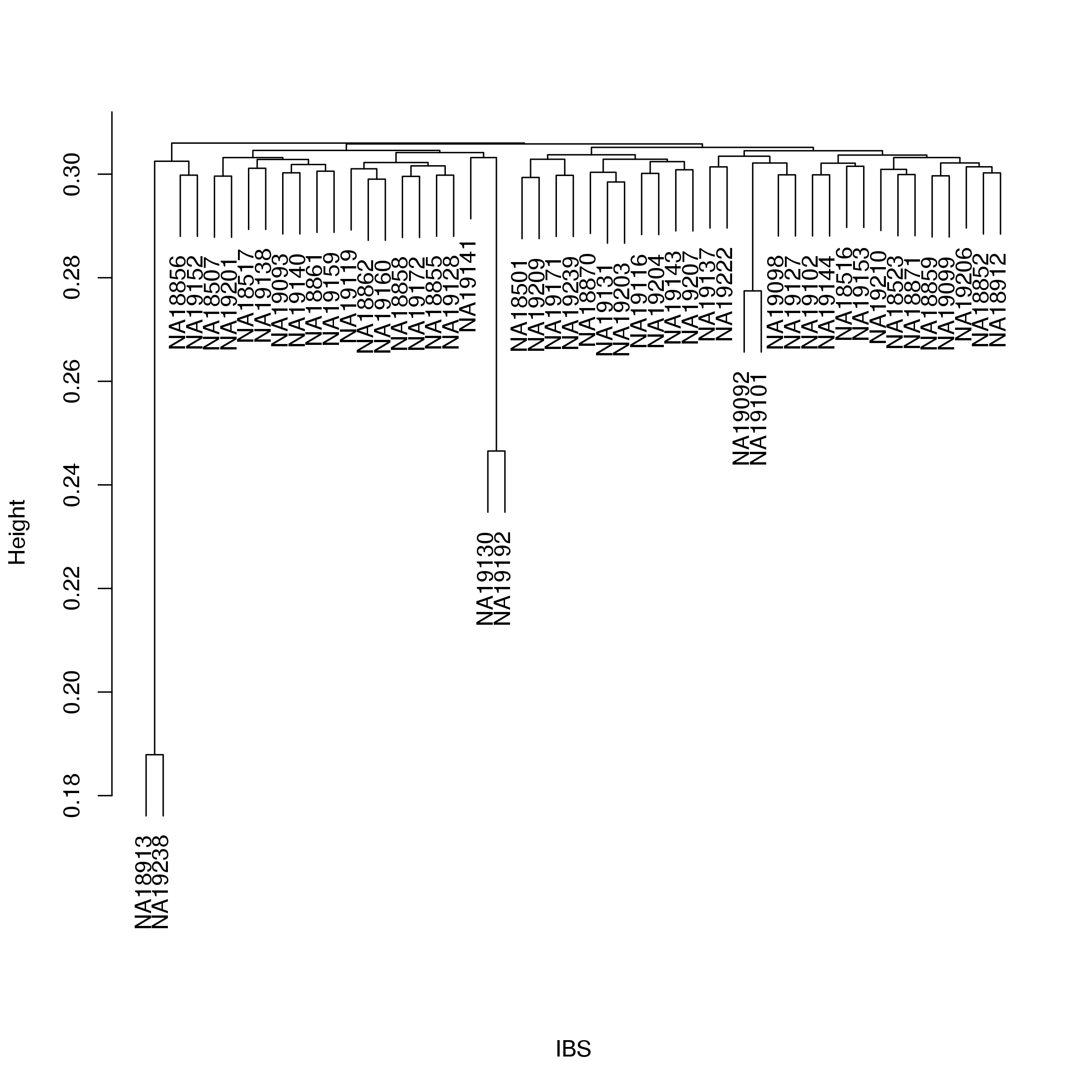
Figure 4: Dendrogram of individuals included in study. Distances are based on identity by state. Three pairs show clear indications of increased relatedness.
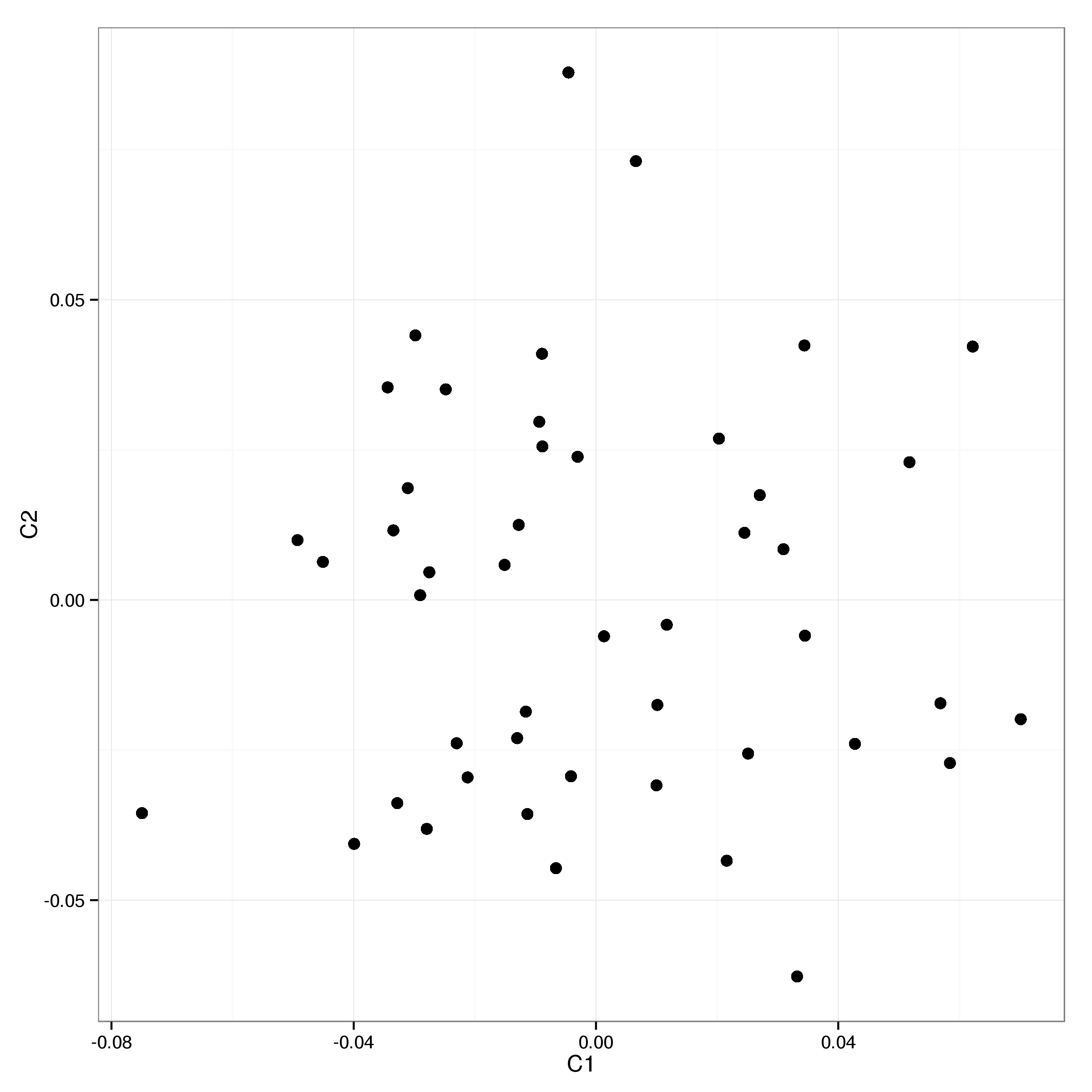
Figure 5: MDS plot of samples remaining after genotype QC.
Plotting the first two MDS components of the remaining 47 samples shows no further evidence for outliers (Figure 5).
Normalising gene expression estimates
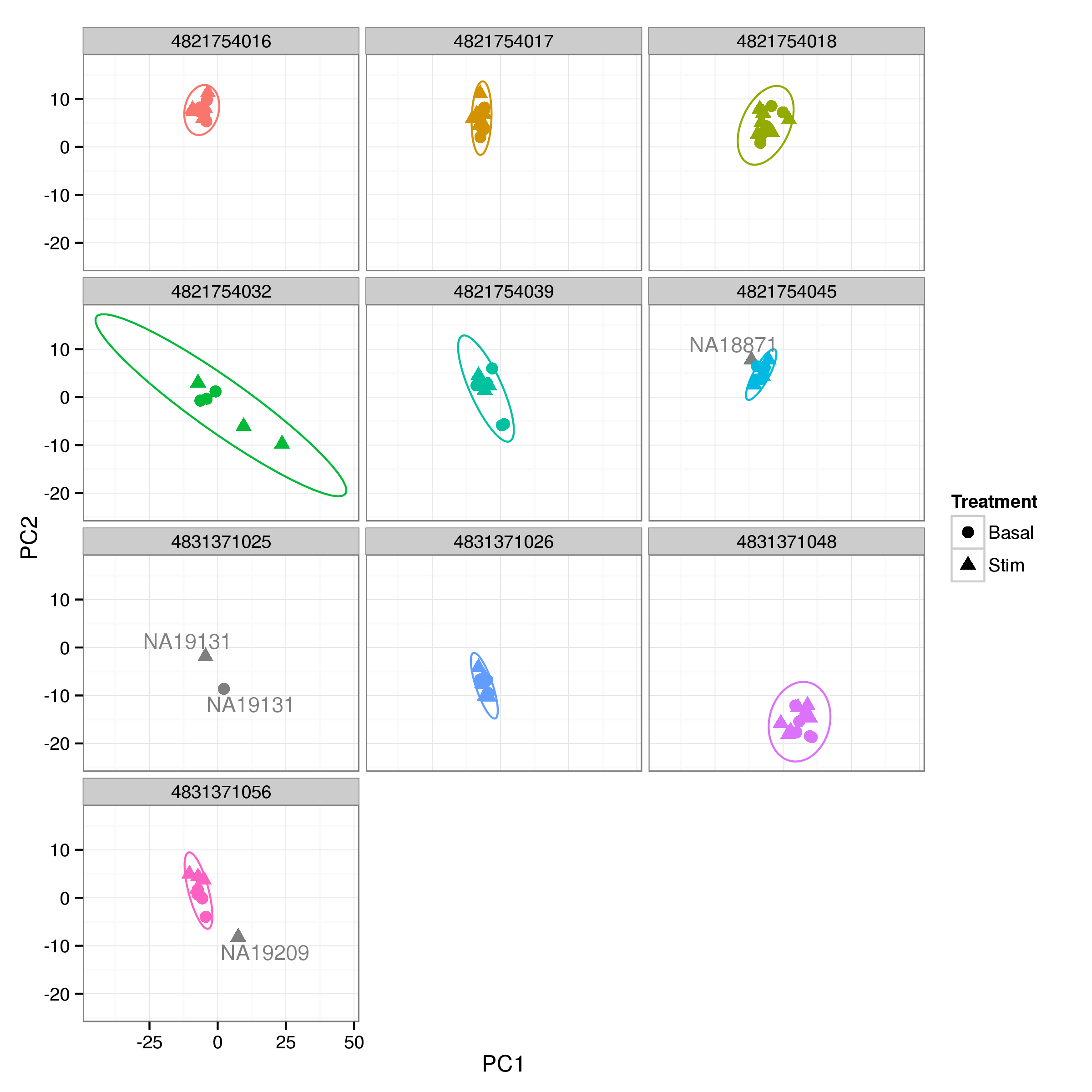
Figure 6: PCA plot of normalised gene expression estimates by sample. Each panal is comprised of the remaining samples for a singel BeadChip. For each BeadChip a 99% confidence ellipse is shown. While the majority of samples cluster well within chips some clear ouliers are visible. Variability between chips is relatively high with chips 563403730 and 563403752 appearing distinct from the bulk of the samples.
Probe intensities were normalised with VSN (Huber et al. 2002). A PCA plot of the initial VSN normalised data (Figure 6) shows clear evidence of samples clustering by BeadChip, as well as 2 obvious outliers, which were removed. In addition the few remaining samples from chip 4831371025 were removed as the entire chip appears to be suspect.
The remaining samples were normalised separately for each BeadChip and differences between groups corrected with ComBat (Johnson, Li, and Rabinovic 2007; Leek et al. 2015), preserving differences due to heat shock stimulation.
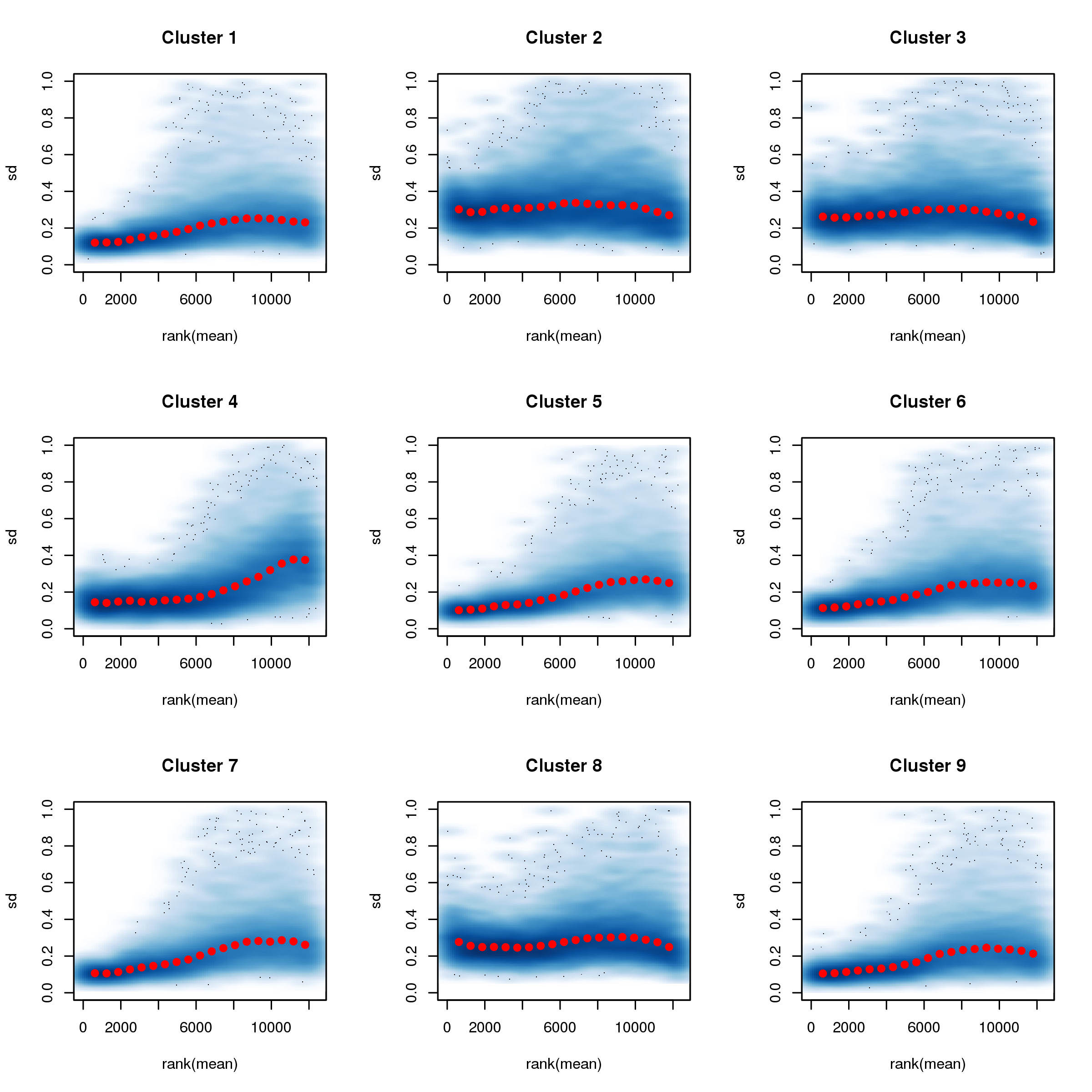
Figure 7: VSN fits to grouped dataset.
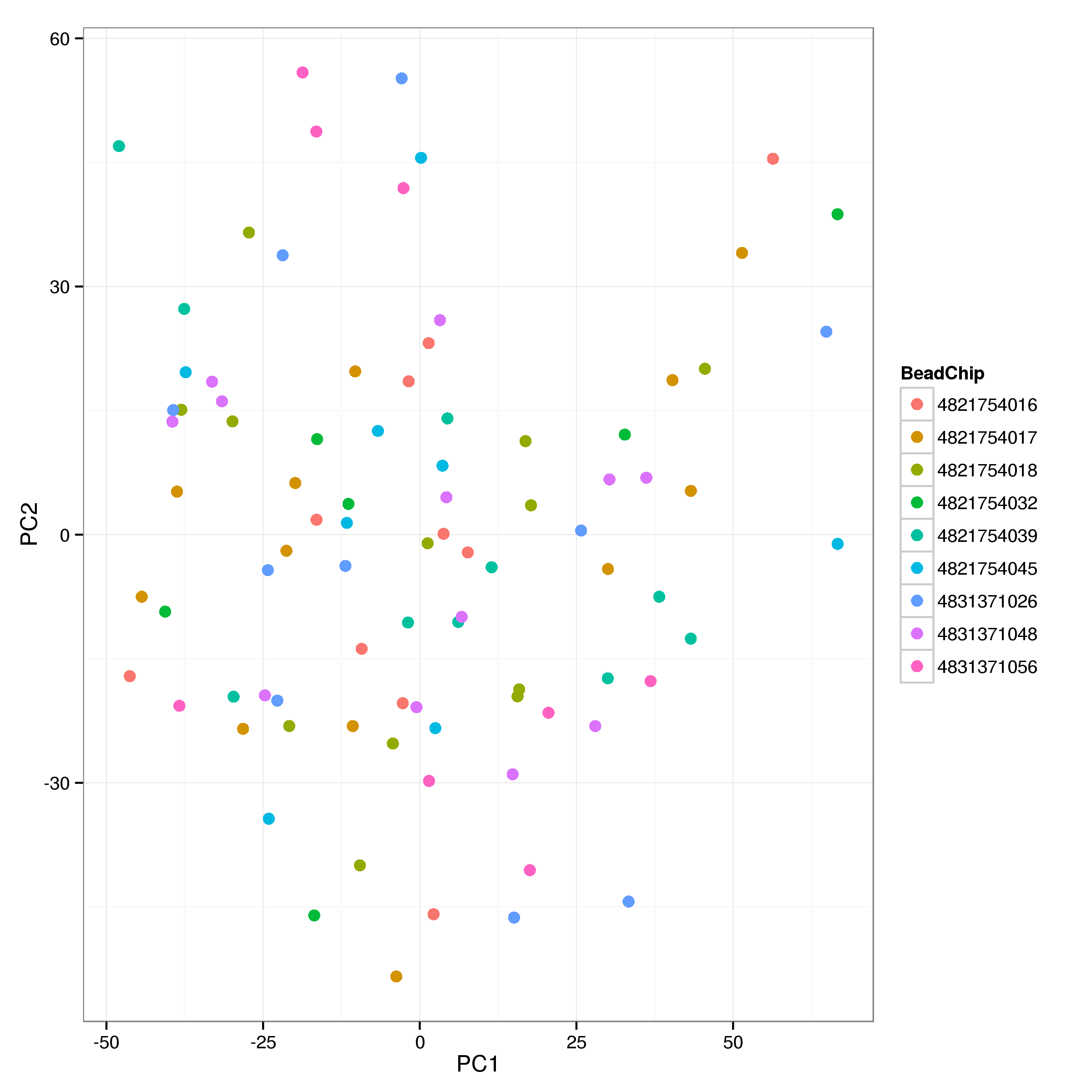
Figure 8: PCA plot of ComBat corrected gene expression.
Differential expression analysis
All remaining samples were analysed for differences in gene expression between the basal stimulated states, pairing samples from the same individual, using the limma Bioconductor package (M. E. Ritchie et al. 2015).
Individual probes were associated with corresponding genes using the updated annotations provided by the illuminaHumanv3.db Bioconductor package (Barbosa-Morais et al. 2010).
Gene N4BP2L2 is associated with two different probes that show evidence of differential expression. One appears to be significantly up regulated while the other is down regulated following heat shock (Table 3). We note that this is the only instance in which multiple probes for a differentially expressed gene provide conflicting evidence in this way. While it is possible that these probes measure two different transcripts that exhibit opposing responses to heat shock we chose to exclude these probes from further analysis to avoid confusion.
GO enrichment analysis
To further investigate the patterns of differential expression a GO enrichment analysis was carried out using the Bioconductor package topGO (Alexa and Rahnenfuhrer 2010). Fisher’s Exact Test was used to determine enrichment separately for significantly (FDR < 0.01) up (Table 4) and down (Table 5) regulated genes.
Gene annotions
In addition to identifying common themes amongst differentially expressed genes the question arises to what extend these genes have been previously associated with the heat shock or, more generally, stress response. We use the set of genes previously linked directly to heat shock (Richter, Haslbeck, and Buchner 2010) and from this create an extended set based on GO terms and PubMed articles linking differentially expressed genes to heat shock response and closely related processes.
Gene Ontology terms
As a first step in highlighting genes not previously known to play a role in this context we identify all significantly up regulated genes that lack GO annotations of obvious relevance to heat shock response. In addition to terms related to stress response and protein folding we also include terms related to cell death and proliferation. To account for the presence of EBV in these cells we ignore all genes annotated with terms related to viral infections. Finally, any remaining genes related to regulation of gene expression are considered to be explained by the large scale changes in gene expression that are taking place in response to heat shock.
PubMed
All genes not annotated with obvious GO terms were subjected to a PubMed search to find publications that link the gene to heat shock or stress response.
Heat shock factor binding
To further establish links between differentially expressed genes and heat shock response we consider the evidence for binding of HSF1 and HSF2 in the promoter regions of these genes. Using binding sites derived fro ChIP-seq data obtained from the K562 cell line (Vihervaara et al. 2013) we annotated our list of differentially expressed genes by cross-referencing it with the list HSF binding genes. Groups of genes corresponding to up or down regulated genes as well as those with existing heat shock related annotations and those without were tested for enrichment of HSF binding genes using Fisher’s exact test.
In addition to the direct evidence from the ChIP-seq data we carried out a scan for the presence of HSF binding motifs in the promoter region (1200bp upstream - 300bp downstream of the TSS) of differentially expressed genes. The scan was based on the PWM defined by SwissRegulon (Pachkov et al. 2007) and carried out with the Bioconductor package PWMEnrich (Stojnic and Diez 2014).
Global heat shock response
To assess the global difference in gene expression induced by heat shock we carried out partial least squares regression (PLS), using the treatment state (basal or stimulated) as a binary response variable and all gene expression probes that passed QC as explanatory variables.
To explore to what extend this response is modulated by genetic variation we used the length and direction of the response vector, i.e. the vector between the basal and stimulated sample for each individual in the space spanned by the first two PLS components, together with the location of the basal sample in the same space as a multivariate phenotype. This was then tested for association with genotypes for SNPs close to differentially expressed genes and their upstream regulators (identified via Ingenuity Pathway Analysis) using the MultiPhen R package (O’Reilly 2012). The GRCh37 coordinates for SNPs were obtained via the SNPlocs.Hsapiens.dbSNP142.GRCh37 Bioconductor package (Pagès 2014) and gene coordinates via the TxDb.Hsapiens.UCSC.hg19.knownGene package (Carlson 2015).
Significance of the observed associations was assessed through a permutation test to account for the structure inherent to the data.
To this end the observed global response phenotype for each individual and covariates used in the model were randomly assigned to one of the observed set of genotypes 1000 times and p-values for the joint model were computed for each permutation. From these we computed false discovery rates by comparing observed p-values to the empirical distribution of minimum p-values from each permutation.
Following up on the observed associations between SNPs and the global heat shock response we investigated possible associations between these SNPs and differentially expreressed genes implicated in the heat shock response. Here we define the gene set of interest as those genes that were significantly up regulated (FDR < 0.01 and fold change > 1.2) following heat shock and that were found to have HSF1 or HSF2 ChIP-seq peaks or binding motifs (P < 0.05) in their promoter region.
Interpreting the PLS components
Further considering the two PLS components it is apparent that the first component has a strong correspondence with the heat shock response, i.e. it separates basal from stimulated samples, whereas the interpretation of the second component is less obvious. We note that treated samples exhibit increased variance in the second component compared to basal samples. We note that the second component does reveal some structure, presenting three distinct clusters, within the stimulated samples but it is not immediately obvious what is underlying this grouping. In an attempted ti elucidate the drivers behind this structure we first assigned each stimulated sample to one of three clusters and then considered patterns of differential expression between them.
Results
Differential expression analysis
In total 1549 probes are identified as differentially expressed at an FDR threshold of 0.01 (see Table 6 for the top 20 results). Of these 701 are up and 848 down regulated following heat shock treatment. Further restricting this list by excluding all probes with a fold change of less than 1.2 leaves 500 probes with 249 and 251 up and down regulated respectively. Figure 9 shows a smoothed volcano plot highlighting probes of particular interest.
| NuID | SymbolReannotated | logFC | AveExpr | t | P.Value | adj.P.Val | B |
|---|---|---|---|---|---|---|---|
| Tiuh76h0KH_ee.1ztM | HSPA1B | 4.47 | 11.9 | 53.9 | 1.12e-52 | 1.39e-48 | 106 |
| xT97CkN_kRuqfFLpUo | DNAJB1 | 3.66 | 10.9 | 42.2 | 1.82e-46 | 1.13e-42 | 93.4 |
| i8lcq61QU7jt6JMo5M | CLK1 | 1.81 | 10.8 | 41.1 | 8.42e-46 | 3.48e-42 | 92 |
| Wivj4OlfbjC0nuHtKk | HSPA6 | 4.2 | 9.35 | 29.9 | 7.83e-38 | 2.43e-34 | 75.2 |
| EXlCsv9_T.ndT0k7u4 | HSPH1 | 1.23 | 12.7 | 23.3 | 8.16e-32 | 2.03e-28 | 61.9 |
| rlU_JFNUR6eeN_.vio | ZFAND2A | 1.92 | 9.45 | 23.2 | 9.79e-32 | 2.03e-28 | 61.8 |
| fq3hG6IHSKuXSqAgB0 | TNFSF14 | 1.42 | 9.37 | 23 | 1.42e-31 | 2.52e-28 | 61.4 |
| ElrogpXmo5QnoQpUj0 | HSPA6 | 2.76 | 7.78 | 21.9 | 2.33e-30 | 3.61e-27 | 58.7 |
| r3kqEepjH6S4e9xfik | FXR1 | 0.933 | 9.88 | 20.8 | 3.28e-29 | 4.53e-26 | 56.1 |
| Bnl1Unc1QXdUHMAcrk | SERPINH1 | 1.9 | 8.73 | 20.4 | 8.6e-29 | 1.07e-25 | 55.2 |
| B.7mrn_C5fk0qPKJDw | TDG | -0.876 | 10.7 | -20.4 | 1.06e-28 | 1.2e-25 | 55 |
| WXeRTp3eQUvd5NH554 | CCL3 | -1.29 | 11.4 | -19.8 | 5.07e-28 | 5.25e-25 | 53.4 |
| 63dUgevknAEiI3h56c | KIAA0907 | 0.94 | 9.49 | 19.5 | 1.04e-27 | 9.92e-25 | 52.7 |
| rsFGq1N.xN6xEQSGdI | HSPA4L | 0.917 | 9.31 | 19.2 | 2.22e-27 | 1.97e-24 | 52 |
| WqK.uImKeJcSOB396U | JUN | 1.22 | 9.22 | 19 | 3.51e-27 | 2.9e-24 | 51.5 |
| raspt5na.ip2mar9e8 | CACYBP | 0.911 | 11.4 | 18.9 | 5.66e-27 | 4.25e-24 | 51.1 |
| iRfeA55zKnrN7AIM14 | DNAJB4 | 1.28 | 7.16 | 18.9 | 5.82e-27 | 4.25e-24 | 51 |
| uUnkaFK9WQBPlJy4pM | DNAJA4 | 1.88 | 8.83 | 18.6 | 1.18e-26 | 8.17e-24 | 50.3 |
| 6qPyUIkSH6PX3tXVRc | IER5 | 1.39 | 10.5 | 18.1 | 4.29e-26 | 2.81e-23 | 49.1 |
| Qt4iQSFdNN7l6CL0e0 | LMAN2L | 0.769 | 8.37 | 18.1 | 4.94e-26 | 3.07e-23 | 48.9 |
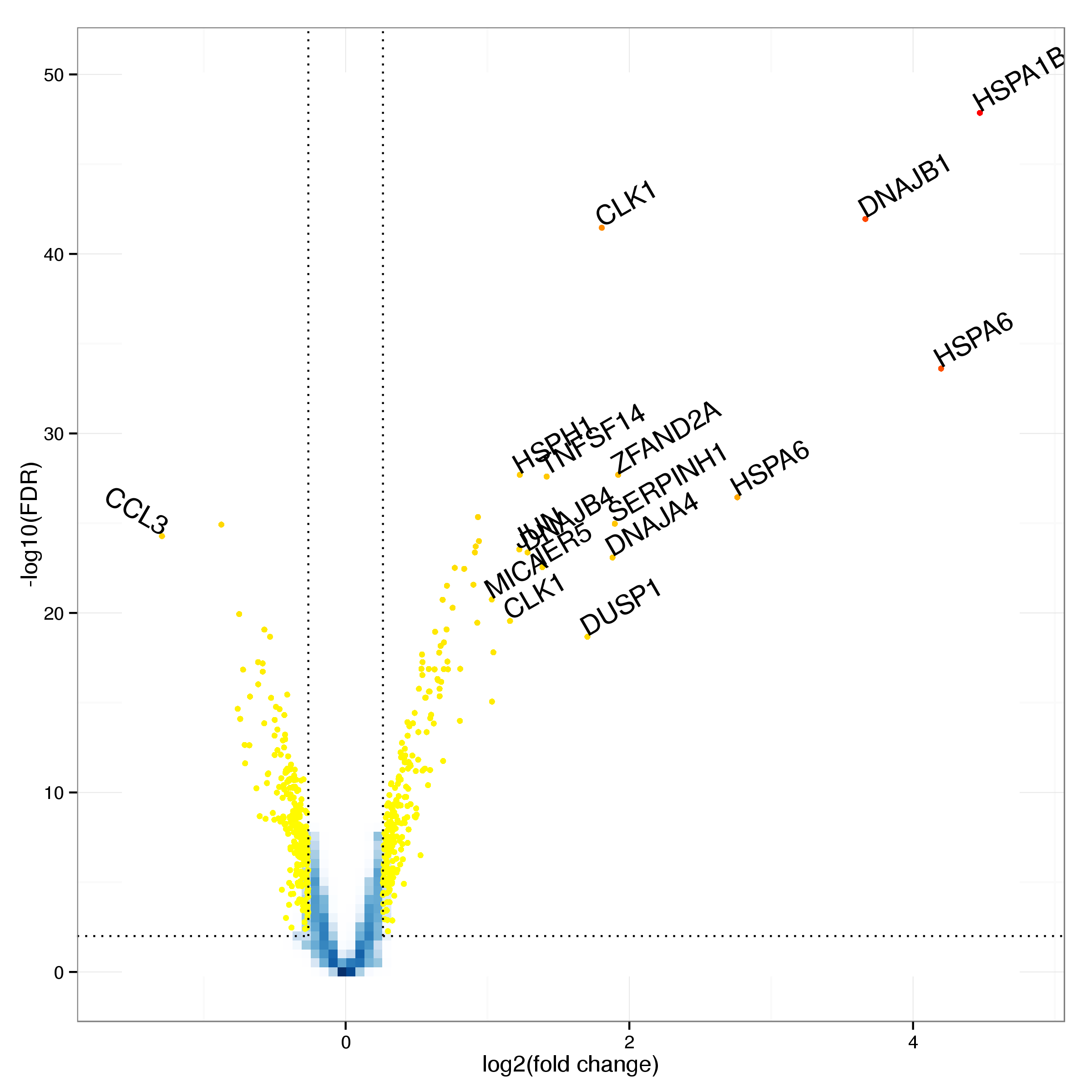
Figure 9: Volcano plot of differential expression results. Probes with an adjusted p-value below 0.01 and a log fold cange of at least 0.5 are shown as yellow and red dots. Probes showing particularly strong evidence of changes in gene expression through a combination of p-value and fold change are labelled with the corresponding gene symbol.
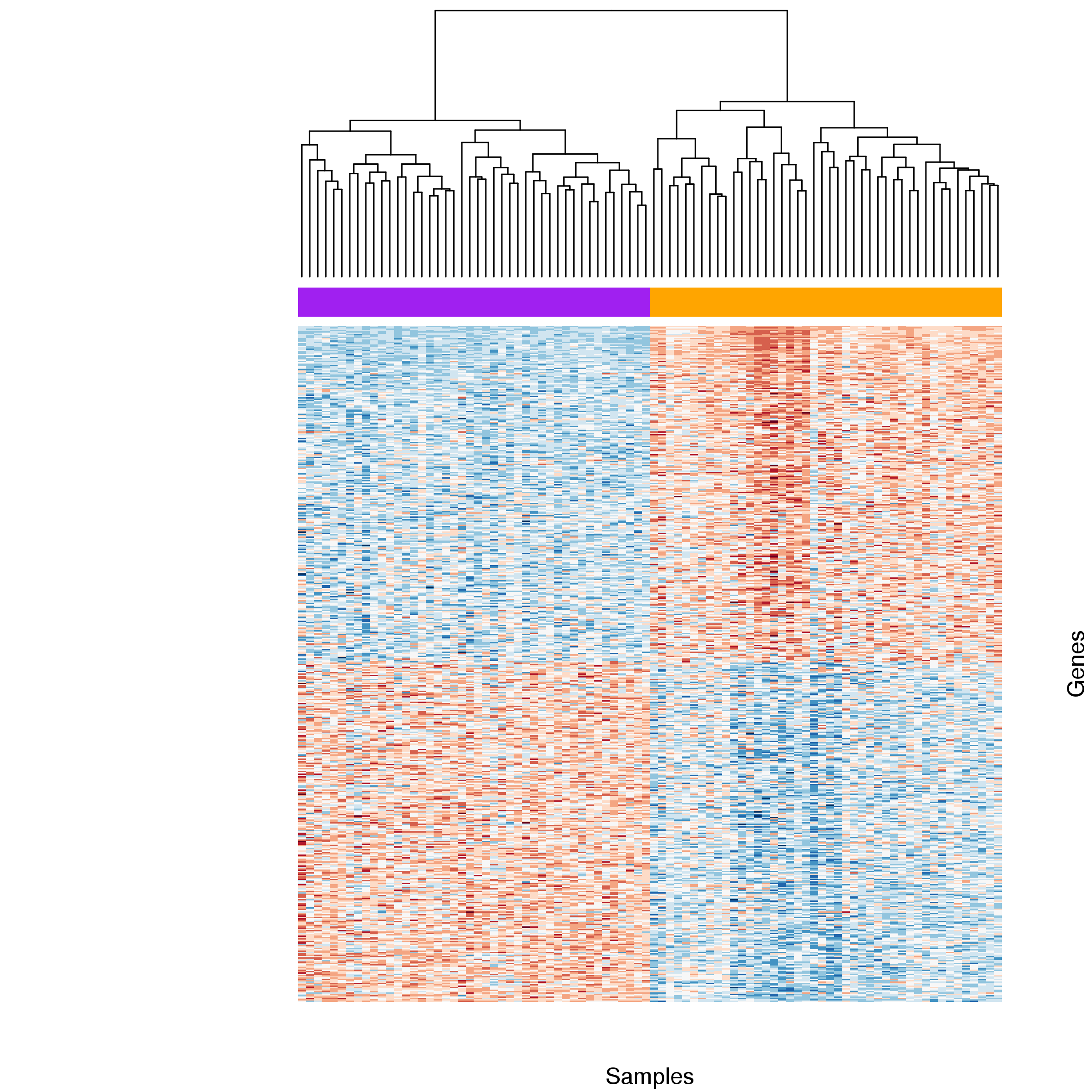
Figure 10: Heatmap comparing gene expression for differentially expressed genes between basal and stimulated samples. Samples were clustered by gene with stimulated (orange) and basal (purple) samples forming two distinct groups. Expression estimates for each gene were scaled and centred across samples. Blue cells correspond to lower and red cells to higher than average expression.
GO enrichment analysis
The GO analysis highlights several categories that are significantly enriched among the up and down regulated genes (7 and 0 categories with an FDR < 0.05 respectively). Considering the top categories (Table 4 and Table 5) it appears that genes up regulated in response to heat shock are predominantly related to stress response (including response to heat [GO:0009408]) and protein folding (including response to unfolded protein [GO:0006986] and protein refolding [GO:0042026]) whereas the leading GO categories for down regulated genes broadly relate to cell proliferation and normal lymphocyte function and include positive regulation of cell cycle [GO:0045787], spindle assembly [GO:0051225] and chemokine production [GO:0032602] but are not sufficiently enriched to be considered significant.
| GO.ID | Term | Annotated | Significant | Expected | Rank in down | up FDR | down FDR |
|---|---|---|---|---|---|---|---|
| GO:0009408 | response to heat | 70 | 13 | 1.5 | 2483 | 0.000226 | 1 |
| GO:0006986 | response to unfolded protein | 97 | 15 | 2.09 | 2753 | 0.000273 | 1 |
| GO:0006457 | protein folding | 133 | 17 | 2.86 | 1593 | 0.00064 | 1 |
| GO:0035966 | response to topologically incorrect prot… | 106 | 15 | 2.28 | 2362 | 0.000939 | 1 |
| GO:0009266 | response to temperature stimulus | 96 | 13 | 2.06 | 2257 | 0.00981 | 1 |
| GO:0042026 | protein refolding | 11 | 5 | 0.24 | 2754 | 0.0277 | 1 |
| GO:0034605 | cellular response to heat | 50 | 9 | 1.07 | 2755 | 0.035 | 1 |
| GO:0043618 | regulation of transcription from RNA pol… | 35 | 7 | 0.75 | 1904 | 0.179 | 1 |
| GO:1900034 | regulation of cellular response to heat | 26 | 6 | 0.56 | 2756 | 0.273 | 1 |
| GO:0043620 | regulation of DNA-templated transcriptio… | 39 | 7 | 0.84 | 2008 | 0.367 | 1 |
| GO:0090083 | regulation of inclusion body assembly | 10 | 4 | 0.21 | 2757 | 0.469 | 1 |
| GO:0034976 | response to endoplasmic reticulum stress | 124 | 12 | 2.67 | 2687 | 0.724 | 1 |
| GO:0042542 | response to hydrogen peroxide | 58 | 8 | 1.25 | 2758 | 0.809 | 1 |
| GO:0080135 | regulation of cellular response to stres… | 303 | 20 | 6.51 | 1467 | 1 | 1 |
| GO:0043066 | negative regulation of apoptotic process | 425 | 25 | 9.14 | 1267 | 1 | 1 |
| GO:0070841 | inclusion body assembly | 13 | 4 | 0.28 | 2759 | 1 | 1 |
| GO:0043069 | negative regulation of programmed cell d… | 428 | 25 | 9.2 | 1299 | 1 | 1 |
| GO:0060548 | negative regulation of cell death | 459 | 26 | 9.87 | 563 | 1 | 1 |
| GO:0036003 | positive regulation of transcription fro… | 14 | 4 | 0.3 | 2760 | 1 | 1 |
| GO:1902175 | regulation of oxidative stress-induced i… | 15 | 4 | 0.32 | 2761 | 1 | 1 |
| GO.ID | Term | Annotated | Significant | Expected | Rank in up | up FDR | down FDR |
|---|---|---|---|---|---|---|---|
| GO:0051225 | spindle assembly | 37 | 6 | 0.82 | 2723 | 1 | 1 |
| GO:0043207 | response to external biotic stimulus | 342 | 20 | 7.56 | 1917 | 1 | 1 |
| GO:0051707 | response to other organism | 342 | 20 | 7.56 | 1918 | 1 | 1 |
| GO:0007049 | cell cycle | 1037 | 45 | 22.9 | 2046 | 1 | 1 |
| GO:0045931 | positive regulation of mitotic cell cycl… | 64 | 7 | 1.41 | 2724 | 1 | 1 |
| GO:0007143 | female meiotic division | 10 | 3 | 0.22 | 2725 | 1 | 1 |
| GO:0009607 | response to biotic stimulus | 355 | 20 | 7.85 | 1711 | 1 | 1 |
| GO:0032496 | response to lipopolysaccharide | 128 | 10 | 2.83 | 1582 | 1 | 1 |
| GO:1903047 | mitotic cell cycle process | 552 | 27 | 12.2 | 2251 | 1 | 1 |
| GO:0008219 | cell death | 1022 | 43 | 22.6 | 44 | 1 | 1 |
| GO:0002237 | response to molecule of bacterial origin | 134 | 10 | 2.96 | 1662 | 1 | 1 |
| GO:0016265 | death | 1025 | 43 | 22.7 | 36 | 1 | 1 |
| GO:0001819 | positive regulation of cytokine producti… | 180 | 12 | 3.98 | 57 | 1 | 1 |
| GO:0006353 | DNA-templated transcription, termination | 57 | 6 | 1.26 | 1559 | 1 | 1 |
| GO:0000278 | mitotic cell cycle | 627 | 29 | 13.9 | 2236 | 1 | 1 |
| GO:1901654 | response to ketone | 60 | 6 | 1.33 | 688 | 1 | 1 |
| GO:0022402 | cell cycle process | 783 | 34 | 17.3 | 2087 | 1 | 1 |
| GO:0000226 | microtubule cytoskeleton organization | 213 | 13 | 4.71 | 2593 | 1 | 1 |
| GO:0000070 | mitotic sister chromatid segregation | 80 | 7 | 1.77 | 2004 | 1 | 1 |
| GO:0051726 | regulation of cell cycle | 557 | 26 | 12.3 | 1865 | 1 | 1 |
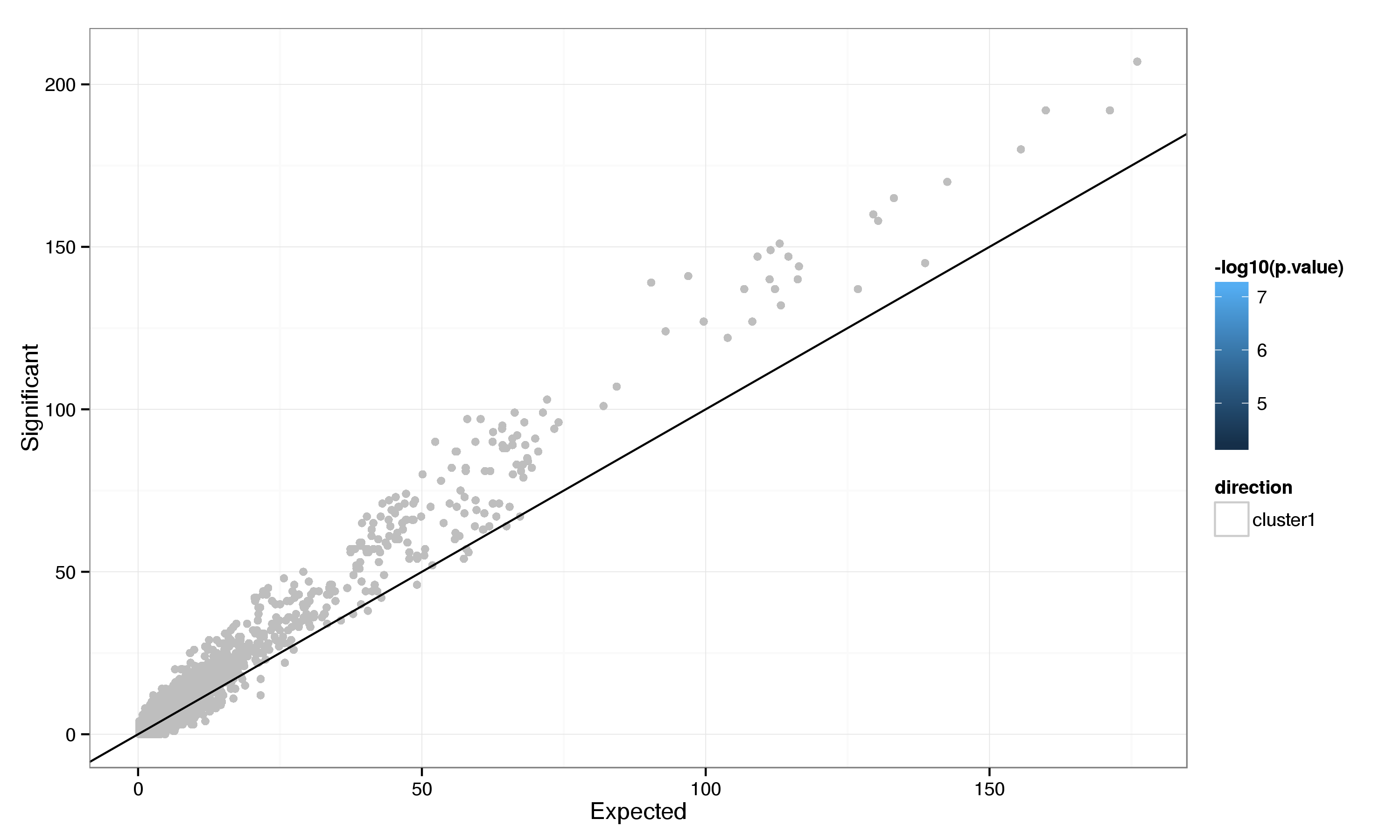
Figure 11: GO category enrichment for differentially expressed genes.
Gene annotations
Of the 226 significantly up regulated genes 24 have been directly linked to the heat shock response and 98 are associated with GO terms that clearly relate to heat shock response (Fisher’s exact test P-value: 2.36e-10) and a further 21 are otherwise linked to the heat shock response in the literature. This leaves 83 genes with no obvious association to heat shock. Using a more inclusive definition of heat shock related processes, also including GO terms related to cell death and proliferation, accounts for an additional 30 genes, leaving 53 genes without any known heatshock association.
| Condition | up | down |
|---|---|---|
| basal HSF1 | 1 (P=4.3e-01, OR= 1.8) | 0 (P=1.00, OR=0.00) |
| basal HSF2 | 7 (P=9.6e-03, OR= 3.2) | 2 (P=0.71, OR=0.81) |
| heat shock HSF1 | 51 (P=4.7e-10, OR= 3.0) | 6 (P=1.00, OR=0.31) |
| heat shock HSF2 | 55 (P=9.4e-09, OR= 2.6) | 10 (P=1.00, OR=0.43) |
| basal HSF1+HSF2 | 1 (P=8.6e-02, OR=14.7) | 0 (P=1.00, OR=0.00) |
| heat shock HSF1+HSF2 | 46 (P=9.1e-15, OR= 4.5) | 4 (P=1.00, OR=0.34) |
There are 7 up-regulated genes without any established role, 8 genes with limited evidence and 29 with substantial evidence for their involvement in the heat shock response that show heat shock factor binding in their promoter region. Further evidence for the binding of HSF1 and HSF2 in these regions is provided by scanning for the presence of the HSF1 binding motif. While this provides less direct evidence than the presence of a ChIP-seq peak and even a good match for the motif is no guarantee for the presence of an active binding site, it enables the detection of potential binding sites that may be inactive in the cell type or state studied through ChIP-seq experiments. Although overall the evidence for the presence of the HSF1 motif is relatively weak, there is a clear increase in the average score for genes that show evidence of HSF binding compared to those that don’t (Figure 12).
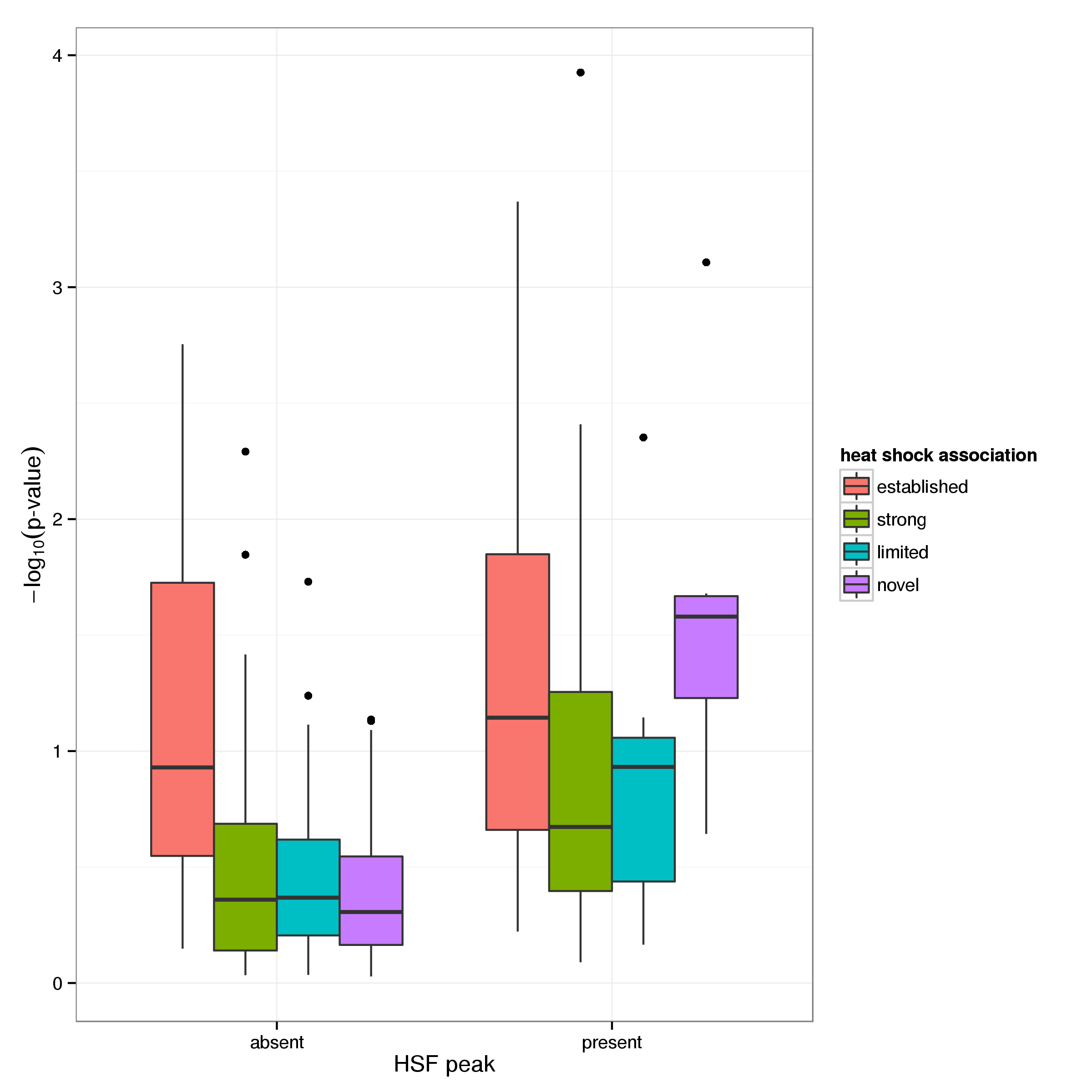
Figure 12: Distribution of p-values for HSF1 motif score.
| expression | ChIPseq | absent | present | frequency |
|---|---|---|---|---|
| up | no | 155 | 9 | 5.49% |
| up | yes | 43 | 17 | 28.33% |
| down | no | 206 | 17 | 7.62% |
| down | yes | 9 | 3 | 25.00% |
| others | no | 7466 | 776 | 9.42% |
| others | yes | 407 | 100 | 19.72% |
Considering the frequency with wich HSF1 motifs occur (using a p-value threshold of 0.05) in the promoter regions of genes with and without ChIP-seq binding evidence (Table 8) it is apparent that the motif is enriched amongst up-regulated genes with corresponding protein binding evidence (Fisher’s exact test p-value: 1.2e-05). This enrichment is less pronounced for down regulated genes (0.071).
Further examining up-regulated genes for evidence of HSF binding reveals that 8 genes not previously characterised as heat shock response genes have ChIP-seq peaks and of these 1 also carry the HSF1 binding motif in their promoter (Table 9 and Table 10).
| ChIPseq | motif | established | limited | novel |
|---|---|---|---|---|
| no | absent | 49 | 85 | 21 |
| no | present | 3 | 5 | 1 |
| yes | absent | 13 | 23 | 7 |
| yes | present | 10 | 6 | 1 |
| Gene Symbol | Fold Change | expression FDR | Motif p-value |
|---|---|---|---|
| NAA16 | 1.57 | 4.77e-17 | 0.683 |
| USPL1 | 1.54 | 1.44e-14 | 0.00444 |
| FAM46A | 1.5 | 3.92e-11 | 0.32 |
| GPBP1 | 1.36 | 1.15e-08 | 0.108 |
| TYW3 | 1.33 | 1.82e-10 | 0.0937 |
| EARS2 | 1.31 | 1.11e-12 | 0.0716 |
| MTCH1 | 1.22 | 5.38e-10 | 0.127 |
| TRMT112 | 1.22 | 4.38e-07 | 0.543 |
Global heat shock response
Considering the outcome of the partial least squares regression it is apparent that there is a considerable change in overall gene expression in response to heat shock. Together the first two PLS components account for 96.1% in the variation observed due to heat shock and provide clear separation of the two treatment groups (Figure 13).
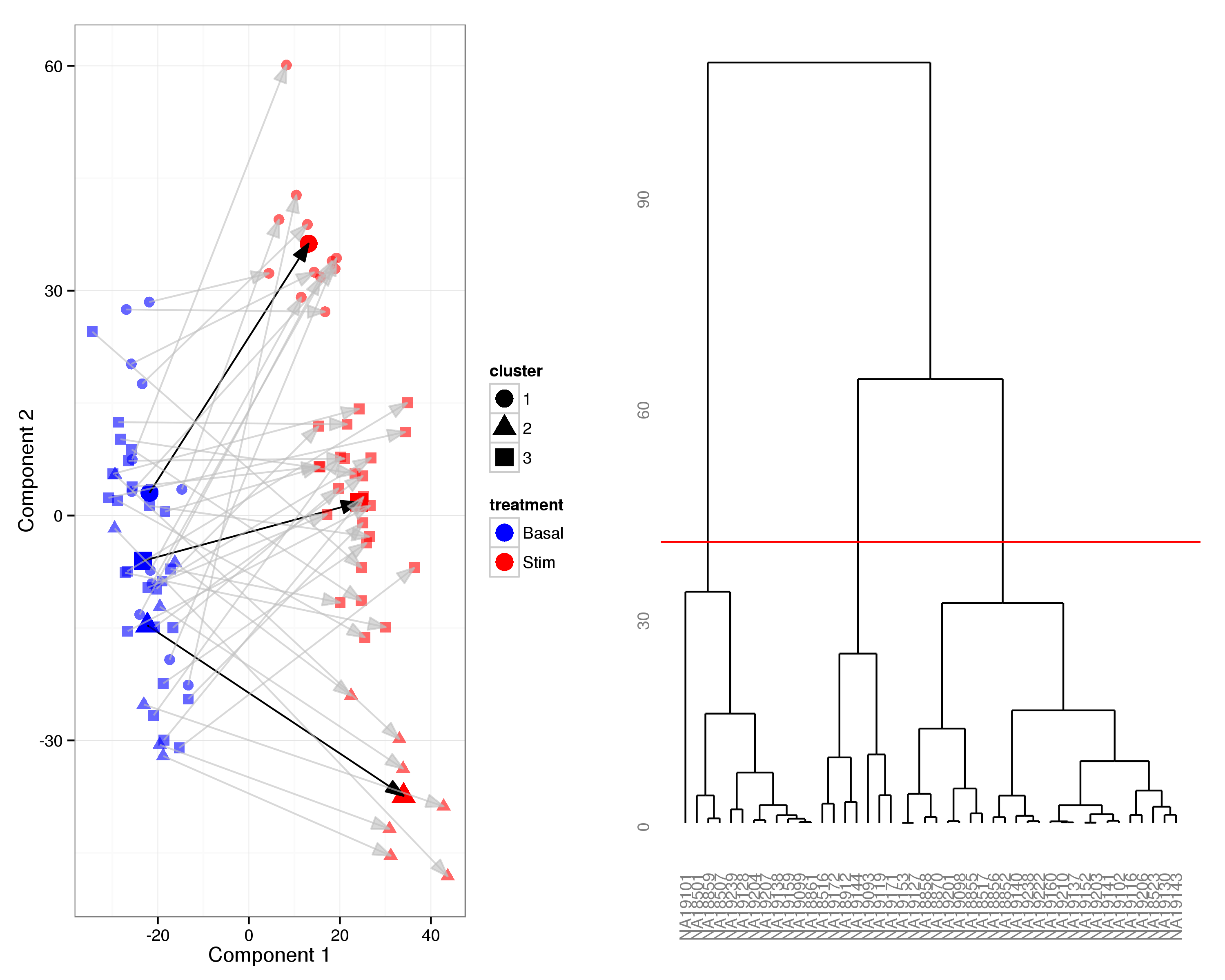
Figure 13: Samples separate clearly by treatment, showing a consistent global effect on gene expression from heat shock. Stimulated samples show evidence of three distinct groups.
Of the 19732 SNPs tested for association with the global response phenotype 2 are considered significant (FDR < 0.05) (Table 11). These SNPs form 2 group of highly correlated genotypes, likely due to local LD structure.
| basal.x | basal.y | direction | distance | Joint model FDR | |
|---|---|---|---|---|---|
| rs10509407 | -4.78 | -4.52 | -0.332 | -4.71 | 0.021 |
| rs12259569 | -4.78 | -4.85 | -0.576 | -4.51 | 0.025 |
| rs12207548 | 1.26 | 0.452 | 1.87 | 0.925 | 0.064 |
| rs7012294 | -0.543 | -2.26 | -2.51 | -3.01 | 0.066 |
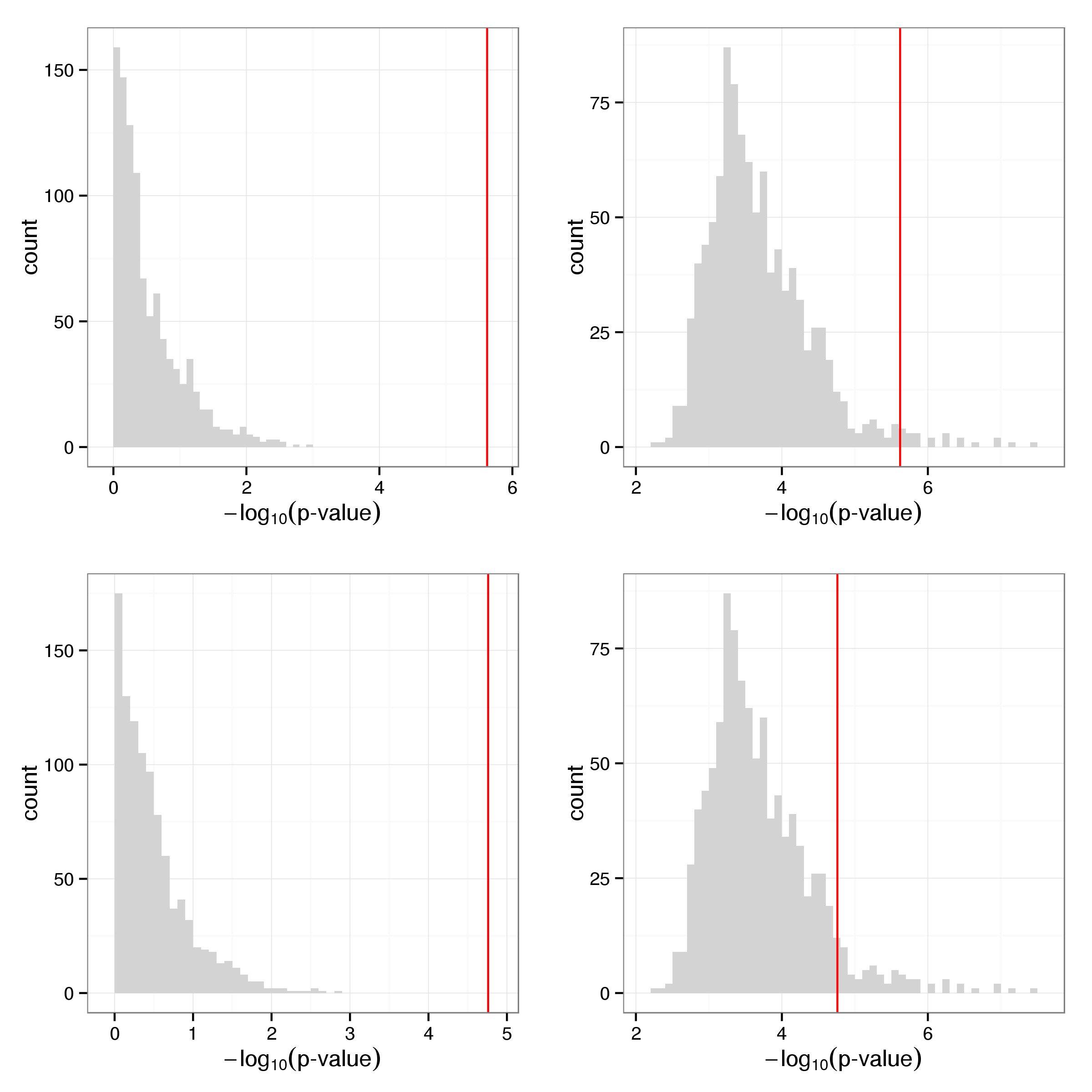
Figure 14: P-values observed after permutation of global response phenotype for SNPs rs10509407 (top) and rs12207548 (bottom). The distribution of p-values for each snp after permutation is shown on the left and the minimum p-value observed in each iteration across all SNPs on the right.
Figure 15 shows two examples of genotypes associating with the global heat shock response. Note that the second one (rs12207548) is located within a CTCF binding site immediately downstream of CDKN1A, an important regulator of cell cycle progression. This SNP is also found to be associated with the expession level of HSF1 in resting lymphocytes (Table 12) It may be interesting to note that allele frequencies vary markedly across populations. While the frequency of the ancestral T allele is 79% in the Yoruba 1000 genomes samples it is only observed with a frequency of 24% in European samples.
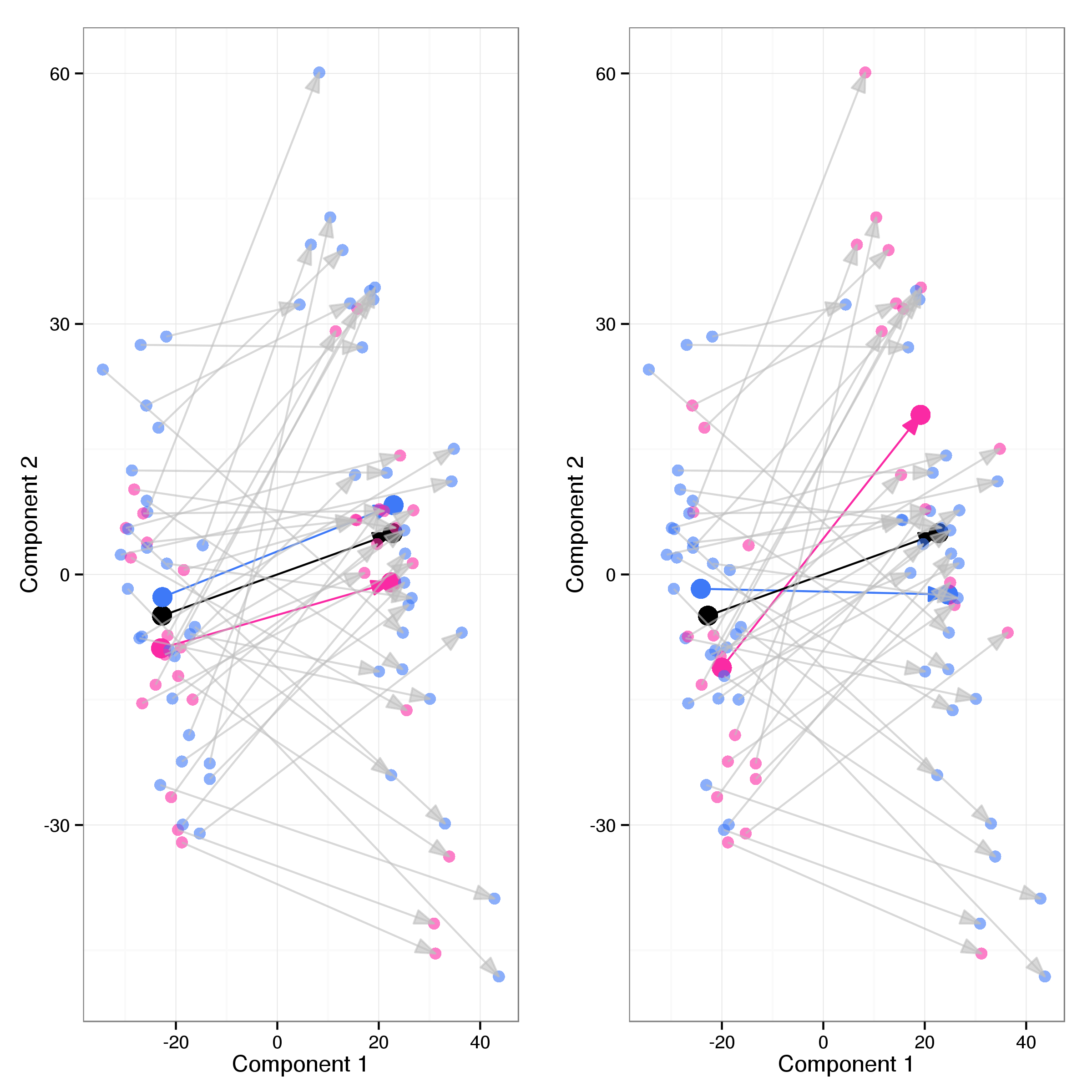
Figure 15: Global response to heat shock by genotype for rs10509407 (left) and rs12207548 (right). Each individual is represented by two points corresponding to basal and stimulated state with arrows connecting paired samples. Genotypes are indicated by colour with blue corresponding to homozyguous carriers of the major allele and red indicating the presence of at least one minor allele.
| SNP | Gene | Statistic | p-value | FDR | beta |
|---|---|---|---|---|---|
| rs12207548 | TNFRSF8 | -5.41 | 3.21e-06 | 0.000751 | -0.477 |
| rs12207548 | HSF1 | -5.35 | 3.84e-06 | 0.000751 | -0.643 |
| rs12207548 | EPHB1 | -5.25 | 5.34e-06 | 0.000751 | -0.532 |
| rs12207548 | SHC1 | -4.72 | 2.92e-05 | 0.00308 | -0.456 |
| rs12207548 | ZC3HAV1 | -4.6 | 4.25e-05 | 0.00358 | -0.399 |
| rs12207548 | ACBD3 | -4.17 | 0.000157 | 0.0102 | -0.279 |
| rs12207548 | UBQLN1 | -4.15 | 0.000169 | 0.0102 | -0.238 |
| rs10509407 | UBQLN1 | 4.07 | 0.000216 | 0.0114 | 0.232 |
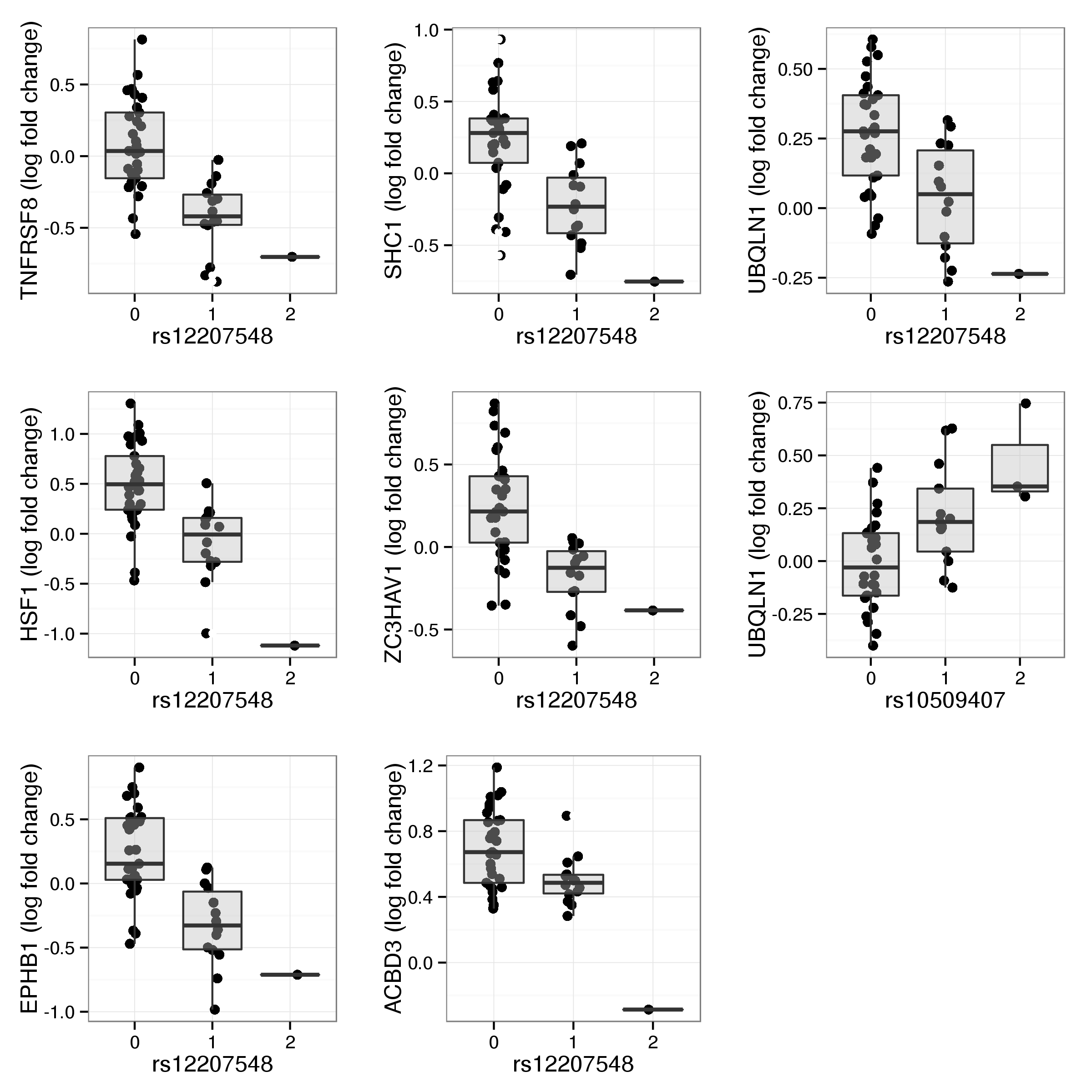
Figure 16: Heat shock response by genotype.
| SNP | Gene | Statistic | p-value | FDR | beta |
|---|---|---|---|---|---|
| rs12207548 | HSPA1B | 3.52 | 0.00111 | 0.467 | 0.304 |
| rs12207548 | PPP5C | 2.8 | 0.00787 | 0.926 | 0.204 |
| rs12207548 | RSRC2 | -2.65 | 0.0116 | 0.926 | -0.166 |
| rs10509407 | EGR1 | 2.48 | 0.0176 | 0.926 | 0.178 |
| rs10509407 | HSPA4 | 2.46 | 0.0183 | 0.926 | 0.126 |
| rs12207548 | HSPA4 | 2.39 | 0.0218 | 0.926 | 0.153 |
| rs10509407 | FAS | -2.3 | 0.0271 | 0.926 | -0.147 |
| rs10509407 | E2F3 | 2.19 | 0.0342 | 0.926 | 0.114 |
| rs10509407 | UBQLN1 | 2.19 | 0.0345 | 0.926 | 0.0915 |
| rs12207548 | HSF1 | -2.17 | 0.0359 | 0.926 | -0.171 |
| rs12207548 | PRR7 | -2.17 | 0.0362 | 0.926 | -0.177 |
| rs10509407 | MAPK14 | 2.12 | 0.04 | 0.926 | 0.106 |
| rs12207548 | PRKCE | -2.08 | 0.0445 | 0.926 | -0.158 |
| rs12207548 | ZFAND2A | 2.06 | 0.046 | 0.926 | 0.199 |
| rs12207548 | NEDD9 | 2.05 | 0.0472 | 0.926 | 0.141 |
| rs12207548 | SPR | 2.03 | 0.0492 | 0.926 | 0.276 |
| rs12207548 | FAS | 2.02 | 0.0504 | 0.926 | 0.163 |
| rs10509407 | TNFSF14 | -1.95 | 0.0583 | 0.926 | -0.171 |
| rs12207548 | SHC1 | -1.91 | 0.0629 | 0.926 | -0.164 |
| rs12207548 | GPR89A | -1.91 | 0.063 | 0.926 | -0.0985 |
| SNP | Gene | Statistic | p-value | FDR | beta |
|---|---|---|---|---|---|
| rs12207548 | HSF1 | 4.23 | 0.000138 | 0.0582 | 0.341 |
| rs12207548 | FOSL1 | 3.58 | 0.00094 | 0.198 | 0.159 |
| rs10509407 | NR3C1 | -3.37 | 0.0017 | 0.239 | -0.154 |
| rs10509407 | UBQLN1 | -3.14 | 0.00323 | 0.311 | -0.14 |
| rs12207548 | ZC3HAV1 | 3.01 | 0.00461 | 0.311 | 0.203 |
| rs12207548 | SHC1 | 2.96 | 0.00522 | 0.311 | 0.182 |
| rs12207548 | TCP1 | 2.92 | 0.00581 | 0.311 | 0.186 |
| rs12207548 | BANP | -2.86 | 0.00677 | 0.311 | -0.154 |
| rs12207548 | TRAF2 | 2.85 | 0.00686 | 0.311 | 0.162 |
| rs10509407 | MTCH1 | -2.75 | 0.00897 | 0.311 | -0.105 |
| rs10509407 | USPL1 | 2.72 | 0.0096 | 0.311 | 0.119 |
| rs12207548 | GNB1 | -2.68 | 0.0106 | 0.311 | -0.132 |
| rs12207548 | JUN | -2.68 | 0.0107 | 0.311 | -0.266 |
| rs10509407 | PADI4 | 2.68 | 0.0108 | 0.311 | 0.121 |
| rs10509407 | UBQLN1 | 2.67 | 0.0111 | 0.311 | 0.101 |
Noting that the minor allele frequency of rs12207548 varies considerably between populations we thought to investigate wether there is evidence of selection at this locus. To this end we calculated FST between the Yoruban and Central European populations for this SNP and compare it to all SNPs with similar MAF in the Yoruban samples.
This estimates the FST for rs12207548 to be 0.31. Of the 1175115 SNPs tested 10.55% have a more extreme value.
Investigating the differences in gene expression between cluster 1 and 2 of the second PLS component we identified a total of 1096 probes as differentially expressed at an FDR threshold of 0.01 (see Table 15 for the top 20 results). Of these 681 are up and 415 down regulated in cluster 2 compared to expression levels in cluster 1. Almost all of these (1094) have a fold change of at least 1.2. Figure 17 shows a smoothed volcano plot highlighting probes of particular interest.
| NuID | SymbolReannotated | logFC | AveExpr | t | P.Value | adj.P.Val | B |
|---|---|---|---|---|---|---|---|
| Z5UZO6CcrqHT1w95b4 | MED22 | 0.855 | 9.28 | 10.9 | 1.69e-14 | 2.1e-10 | 22.6 |
| ltLUV5VtR.VN12swNM | TOMM40 | 0.93 | 11.9 | 10.2 | 1.42e-13 | 8.84e-10 | 20.5 |
| WV5XUSD6vUKnvUhB5Q | GNAI2 | -1.09 | 10.5 | -9.63 | 8.95e-13 | 3.7e-09 | 18.8 |
| rlHpSl1Skrv870kMCs | ZC3H3 | 0.726 | 8.77 | 8.99 | 7.42e-12 | 1.62e-08 | 16.8 |
| HS7hLlJQ7kiujit_4A | AIMP2 | 0.779 | 11.2 | 8.98 | 7.69e-12 | 1.62e-08 | 16.8 |
| B_gkHi0cdQCrXnUl6c | C1orf112 | 0.903 | 8.81 | 8.98 | 7.81e-12 | 1.62e-08 | 16.7 |
| x7leKemno5ulDACSFw | INF2 | 1.27 | 9.56 | 8.74 | 1.75e-11 | 3.11e-08 | 16 |
| or3hBLFTVUSxV9J0v0 | BCL7B | 0.977 | 8.13 | 8.63 | 2.59e-11 | 4.02e-08 | 15.6 |
| WebhLrV3VaH5H71_.U | PUF60 | 0.967 | 11.2 | 8.42 | 5.25e-11 | 7.24e-08 | 14.9 |
| rOezhdXiRW4eVTn9V8 | RANGAP1 | 1.04 | 10.9 | 8.28 | 8.53e-11 | 9.84e-08 | 14.5 |
| Zhq6dHlPtFK_gd_Xe4 | NOP2 | 0.776 | 11.1 | 8.27 | 8.72e-11 | 9.84e-08 | 14.4 |
| 9Xop_fqHop_foS9d7s | HPS6 | 0.937 | 9.66 | 8.22 | 1.06e-10 | 1.1e-07 | 14.2 |
| xKpnfhHuX0.1JUSXq0 | TYSND1 | 0.88 | 9.6 | 8.18 | 1.19e-10 | 1.13e-07 | 14.1 |
| lEdet6Oqeefi5Iqeaw | SLC25A19 | 1.23 | 8.89 | 8.06 | 1.84e-10 | 1.51e-07 | 13.7 |
| 01OVCvVK5KEiu6XPsU | C17orf53 | 0.982 | 8.75 | 8.06 | 1.84e-10 | 1.51e-07 | 13.7 |
| i15CjPunohOJCAJd6g | LSP1 | -1.44 | 8.31 | -8.04 | 1.95e-10 | 1.51e-07 | 13.7 |
| HzqJNpx567t9VBpGis | CD74 | -1.49 | 8.28 | -7.97 | 2.52e-10 | 1.84e-07 | 13.4 |
| BesiCOkSjoplUIaEuw | E4F1 | 1.08 | 9.03 | 7.91 | 3.11e-10 | 2.15e-07 | 13.2 |
| NnJ1ITq_NFyKJ3pwpE | NPRL3 | 0.949 | 10.1 | 7.81 | 4.35e-10 | 2.75e-07 | 12.9 |
| upeWHh7uk6JOnvVQuQ | SNAPC4 | 0.729 | 8.9 | 7.8 | 4.5e-10 | 2.75e-07 | 12.9 |
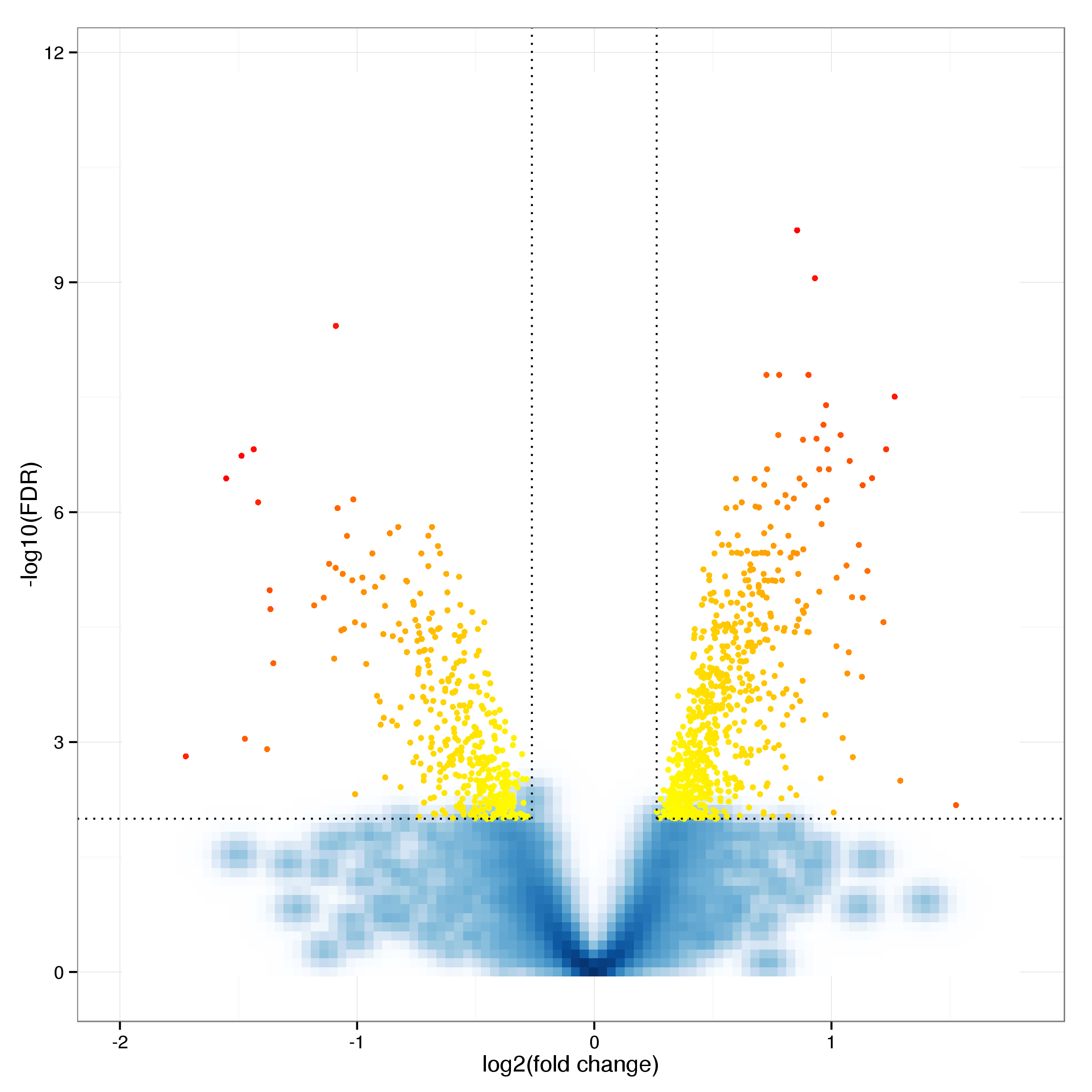
Figure 17: Volcano plot of differential expression results between clusters 1 and 2. Probes with an adjusted p-value below 0.01 and a log fold cange of at least 0.5 are shown as yellow and red dots.
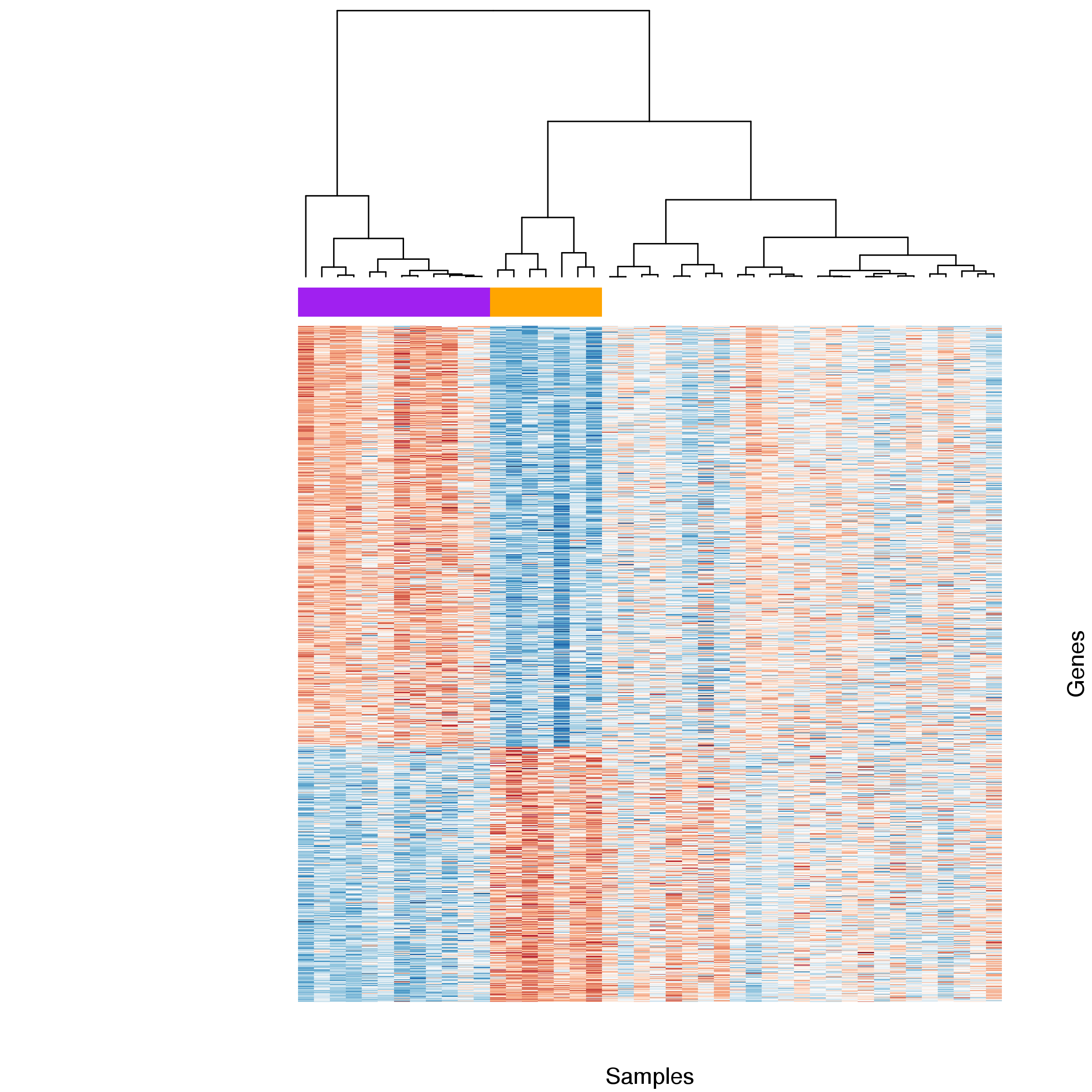
Figure 18: Heatmap comparing gene expression for genes differentially expressed between clusters 1 and 2. Expression estimates for each gene were scaled and centred across samples. Blue cells correspond to lower and red cells to higher than average expression.
To further characterise the differnces in gene expression we carried out a GO enrichment analysis for genes differentially expressed between clusters 1 and 2. This reveals strong enrichment for several GO categories (21 and 6 enriched with FDR < 0.05 in cluster 1 and cluster 2 respectively). Genes with increased expression in cluster 1 (Table 16) are enriched for biological processes related to DNA replication whereas those with increased expression in cluster 2 (Table 17) are annotated with processes related to the heat shock response.
| GO.ID | Term | Annotated | Significant | Expected | Rank in cluster2 | cluster1 FDR | cluster2 FDR |
|---|---|---|---|---|---|---|---|
| GO:0034660 | ncRNA metabolic process | 257 | 51 | 15.8 | 3245 | 1.84e-06 | 1 |
| GO:0034470 | ncRNA processing | 170 | 38 | 10.5 | 3252 | 1.07e-05 | 1 |
| GO:0022613 | ribonucleoprotein complex biogenesis | 198 | 41 | 12.2 | 3249 | 2.69e-05 | 1 |
| GO:0042254 | ribosome biogenesis | 117 | 29 | 7.21 | 3235 | 7.26e-05 | 1 |
| GO:0006310 | DNA recombination | 146 | 33 | 9 | 3159 | 8.96e-05 | 1 |
| GO:0006399 | tRNA metabolic process | 96 | 25 | 5.92 | 3259 | 0.000247 | 1 |
| GO:0006261 | DNA-dependent DNA replication | 81 | 21 | 4.99 | 2991 | 0.00307 | 1 |
| GO:0006271 | DNA strand elongation involved in DNA re… | 25 | 11 | 1.54 | 3260 | 0.00469 | 1 |
| GO:0022616 | DNA strand elongation | 26 | 11 | 1.6 | 3261 | 0.00725 | 1 |
| GO:0006281 | DNA repair | 276 | 45 | 17 | 3183 | 0.00767 | 1 |
| GO:0006260 | DNA replication | 192 | 35 | 11.8 | 2975 | 0.00852 | 1 |
| GO:0006364 | rRNA processing | 86 | 21 | 5.3 | 3205 | 0.00895 | 1 |
| GO:0000278 | mitotic cell cycle | 627 | 82 | 38.7 | 3222 | 0.00937 | 1 |
| GO:0016072 | rRNA metabolic process | 88 | 21 | 5.43 | 3210 | 0.0132 | 1 |
| GO:0006312 | mitotic recombination | 28 | 11 | 1.73 | 3262 | 0.0175 | 1 |
| GO:0031396 | regulation of protein ubiquitination | 134 | 27 | 8.26 | 2639 | 0.0183 | 1 |
| GO:0051340 | regulation of ligase activity | 70 | 18 | 4.32 | 3139 | 0.0213 | 1 |
| GO:0006259 | DNA metabolic process | 532 | 71 | 32.8 | 2681 | 0.023 | 1 |
| GO:1903047 | mitotic cell cycle process | 552 | 72 | 34 | 3157 | 0.0425 | 1 |
| GO:0051438 | regulation of ubiquitin-protein transfer… | 67 | 17 | 4.13 | 3132 | 0.0468 | 1 |
| GO.ID | Term | Annotated | Significant | Expected | Rank in cluster1 | cluster1 FDR | cluster2 FDR |
|---|---|---|---|---|---|---|---|
| GO:0034976 | response to endoplasmic reticulum stress | 124 | 20 | 4.45 | 2591 | 1 | 0.00683 |
| GO:0030968 | endoplasmic reticulum unfolded protein r… | 74 | 15 | 2.66 | 2180 | 1 | 0.00726 |
| GO:0034620 | cellular response to unfolded protein | 74 | 15 | 2.66 | 2181 | 1 | 0.00726 |
| GO:0006986 | response to unfolded protein | 97 | 17 | 3.48 | 2381 | 1 | 0.0128 |
| GO:0035967 | cellular response to topologically incor… | 81 | 15 | 2.91 | 1932 | 1 | 0.0243 |
| GO:0035966 | response to topologically incorrect prot… | 106 | 17 | 3.81 | 1780 | 1 | 0.0427 |
| GO:0032075 | positive regulation of nuclease activity | 49 | 11 | 1.76 | 1838 | 1 | 0.064 |
| GO:0032069 | regulation of nuclease activity | 53 | 11 | 1.9 | 1992 | 1 | 0.141 |
| GO:0006987 | activation of signaling protein activity… | 46 | 10 | 1.65 | 1692 | 1 | 0.213 |
| GO:0080135 | regulation of cellular response to stres… | 303 | 30 | 10.9 | 1060 | 1 | 0.554 |
| GO:1903573 | negative regulation of response to endop… | 18 | 6 | 0.65 | 3374 | 1 | 0.554 |
| GO:0009101 | glycoprotein biosynthetic process | 206 | 22 | 7.4 | 3105 | 1 | 1 |
| GO:0018196 | peptidyl-asparagine modification | 91 | 13 | 3.27 | 3279 | 1 | 1 |
| GO:0018279 | protein N-linked glycosylation via aspar… | 91 | 13 | 3.27 | 3280 | 1 | 1 |
| GO:1900101 | regulation of endoplasmic reticulum unfo… | 15 | 5 | 0.54 | 3375 | 1 | 1 |
| GO:0031344 | regulation of cell projection organizati… | 185 | 20 | 6.65 | 2556 | 1 | 1 |
| GO:1902275 | regulation of chromatin organization | 83 | 12 | 2.98 | 538 | 1 | 1 |
| GO:0006487 | protein N-linked glycosylation | 95 | 13 | 3.41 | 3288 | 1 | 1 |
| GO:0045664 | regulation of neuron differentiation | 201 | 21 | 7.22 | 2316 | 1 | 1 |
| GO:0045860 | positive regulation of protein kinase ac… | 273 | 26 | 9.81 | 3170 | 1 | 1 |
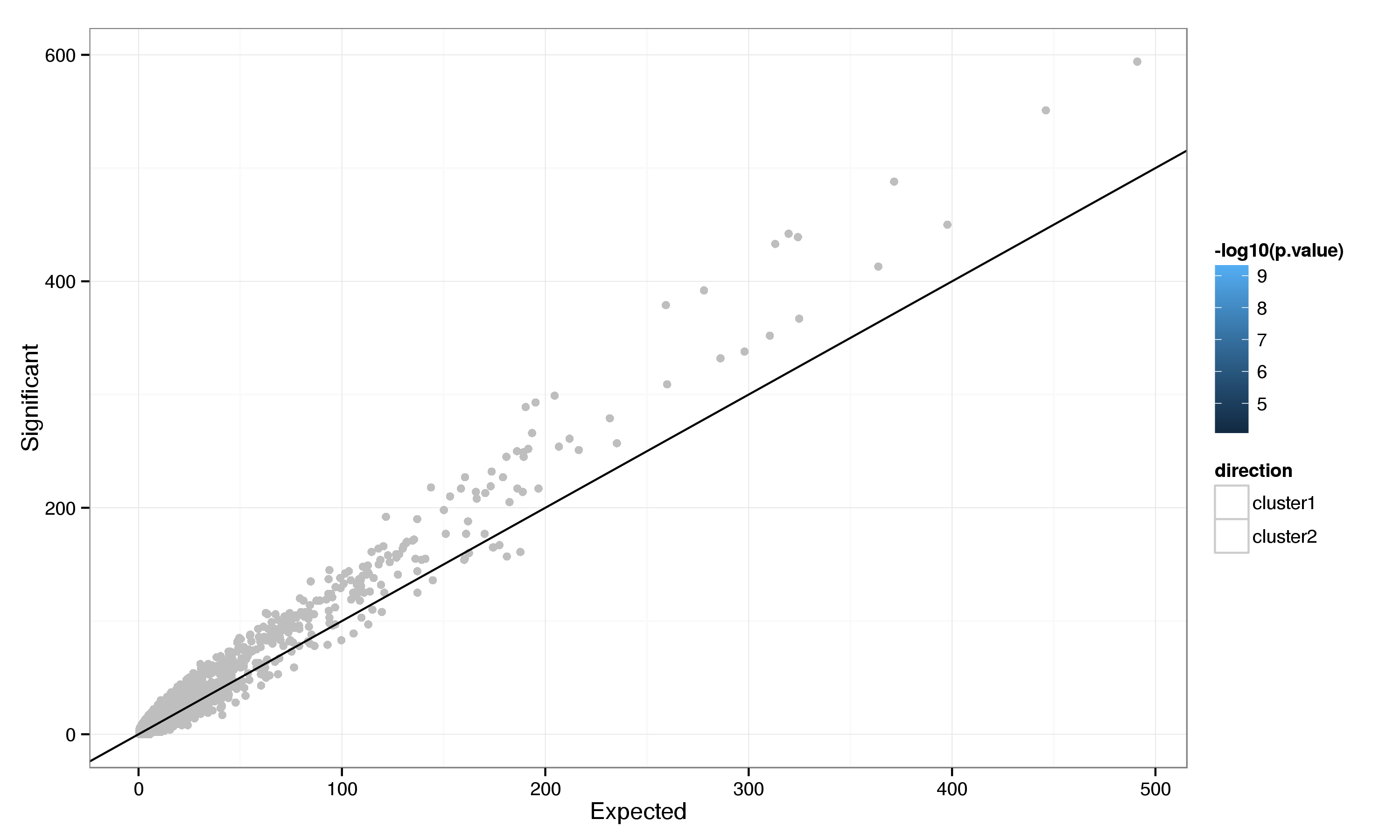
Figure 19: GO category enrichment for genes differentially expressed between PLS clusters.
Appendix
Custom functions used
Session Info
R version 3.2.1 (2015-06-18) Platform: x86_64-pc-linux-gnu (64-bit) Running under: Debian GNU/Linux stretch/sid
locale: [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages: [1] grid stats4 parallel methods stats graphics grDevices [8] utils datasets base
other attached packages: [1] PWMEnrich.Hsapiens.background_4.2.0
[2] PWMEnrich_4.4.0
[3] biomaRt_2.24.0
[4] RISmed_2.1.5
[5] MultiPhen_2.0.1
[6] meta_4.3-0
[7] epitools_0.5-7
[8] abind_1.4-3
[9] plsdepot_0.1.17
[10] MatrixEQTL_2.1.1
[11] plink2R_1.0
[12] RcppEigen_0.3.2.5.0
[13] Rcpp_0.12.0
[14] TxDb.Hsapiens.UCSC.hg19.knownGene_3.1.2 [15] GenomicFeatures_1.20.1
[16] BSgenome.Hsapiens.UCSC.hg19_1.4.0
[17] SNPlocs.Hsapiens.dbSNP142.GRCh37_0.99.5 [18] BSgenome_1.36.2
[19] rtracklayer_1.28.6
[20] Biostrings_2.36.1
[21] XVector_0.8.0
[22] GenomicRanges_1.20.5
[23] topGO_2.20.0
[24] SparseM_1.6
[25] GO.db_3.1.2
[26] graph_1.46.0
[27] ggdendro_0.1-15
[28] reshape2_1.4.1
[29] pander_0.5.2
[30] illuminaHumanv3.db_1.26.0
[31] org.Hs.eg.db_3.1.2
[32] RSQLite_1.0.0
[33] DBI_0.3.1
[34] AnnotationDbi_1.30.1
[35] GenomeInfoDb_1.4.1
[36] IRanges_2.2.5
[37] S4Vectors_0.6.2
[38] stringr_1.0.0
[39] readr_0.1.1.9000
[40] tidyr_0.2.0
[41] dplyr_0.4.2
[42] ggplot2_1.0.1
[43] RColorBrewer_1.1-2
[44] gplots_2.17.0
[45] sva_3.14.0
[46] genefilter_1.50.0
[47] mgcv_1.8-6
[48] nlme_3.1-121
[49] scatterplot3d_0.3-36
[50] limma_3.24.15
[51] vsn_3.36.0
[52] affy_1.46.1
[53] Biobase_2.28.0
[54] BiocGenerics_0.14.0
[55] gdata_2.17.0
[56] proto_0.3-10
[57] MASS_7.3-42
[58] pushoverr_0.1.4
[59] knitrBootstrap_1.0.0
[60] knitr_1.11
[61] rmarkdown_0.7
[62] BiocInstaller_1.18.4
[3] evd_2.3-0 markdown_0.7.7
[5] futile.logger_1.4.1 mice_2.22
[7] affyio_1.36.0 codetools_0.2-11
[9] splines_3.2.1 R.methodsS3_1.7.0
[11] nloptr_1.0.4 Rsamtools_1.20.4
[13] seqLogo_1.34.0 pbkrtest_0.4-2
[15] annotate_1.46.1 R.oo_1.19.0
[17] httr_1.0.0 assertthat_0.1
[19] Matrix_1.2-2 lazyeval_0.1.10
[21] formatR_1.2 quantreg_5.11
[23] htmltools_0.2.6 tools_3.2.1
[25] gtable_0.1.2 preprocessCore_1.30.0
[27] lme4_1.1-8 gtools_3.5.0
[29] XML_3.98-1.3 zlibbioc_1.14.0
[31] scales_0.2.5 lambda.r_1.1.7
[33] HardyWeinberg_1.5.5 yaml_2.1.13
[35] rpart_4.1-10 stringi_0.5-5
[37] randomForest_4.6-10 caTools_1.17.1
[39] BiocParallel_1.2.9 bitops_1.0-6
[41] evaluate_0.7.2 lattice_0.20-31
[43] GenomicAlignments_1.4.1 labeling_0.3
[45] plyr_1.8.3 magrittr_1.5
[47] R6_2.1.0 sp_1.1-1
[49] survival_2.38-3 RCurl_1.95-4.7
[51] nnet_7.3-10 car_2.0-25
[53] futile.options_1.0.0 KernSmooth_2.23-15
[55] digest_0.6.8 xtable_1.7-4
[57] R.utils_2.1.0 munsell_0.4.2
References
Alexa, Adrian, and Jorg Rahnenfuhrer. 2010. TopGO: TopGO: Enrichment Analysis for Gene Ontology.
Barbosa-Morais, Nuno L., Mark J. Dunning, Shamith A. Samarajiwa, Jeremy F. J. Darot, Matthew E. Ritchie, Andy G. Lynch, and Simon Tavaré. 2010. “A Re-Annotation Pipeline for Illumina BeadArrays: Improving the Interpretation of Gene Expression Data.” Nucleic Acids Research 38 (3): e17. doi:10.1093/nar/gkp942.
Carlson, Marc. 2015. TxDb.Hsapiens.UCSC.hg19.knownGene: Annotation Package for TxDb Object(s).
Chang, Christopher, Carson Chow, Laurent Tellier, Shashaank Vattikuti, Shaun Purcell, and James Lee. 2015. “Second-Generation PLINK: Rising to the Challenge of Larger and Richer Datasets.” GigaScience 4 (1): 7. doi:10.1186/s13742-015-0047-8.
Gibbs, Richard et al. 2010. “The International HapMap Project.” Nature 426: 789–96.
Huber, Wolfgang, Anja von Heydebreck, Holger Sueltmann, Annemarie Poustka, and Martin Vingron. 2002. “Variance Stabilization Applied to Microarray Data Calibration and to the Quantification of Differential Expression.” Bioinformatics 18 Suppl. 1: S96–S104.
Johnson, W. Evan, Cheng Li, and Ariel Rabinovic. 2007. “Adjusting Batch Effects in Microarray Expression Data Using Empirical Bayes Methods.” Biostatistics 8 (1): 118–27. doi:10.1093/biostatistics/kxj037.
Kee, C., K. Y. Cheong, K. Pham, G. W. Waterer, and S. E. L. Temple. 2008. “Genetic Variation in Heat Shock Protein 70 Is Associated with Septic Shock: Narrowing the Association to a Specific Haplotype.” International Journal of Immunogenetics 35 (6). Blackwell Publishing Ltd: 465–73. doi:10.1111/j.1744-313X.2008.00812.x.
Leek, Jeffrey T., W. Evan Johnson, Hilary S. Parker, Elana J. Fertig, Andrew E. Jaffe, and John D. Storey. 2015. Sva: Surrogate Variable Analysis.
Martin, Annalise M., David Nolan, Silvana Gaudieri, Coral Ann Almeida, Richard Nolan, Ian James, Filipa Carvalho, et al. 2004. “Predisposition to Abacavir Hypersensitivity Conferred by HLA-B*5701 and a Haplotypic Hsp70-Hom Variant.” Proceedings of the National Academy of Sciences 101 (12): 4180–85. doi:10.1073/pnas.0307067101.
O’Reilly, Clive J. AND Pomyen, Paul F. AND Hoggart. 2012. “MultiPhen: Joint Model of Multiple Phenotypes Can Increase Discovery in GWAS.” PLoS ONE 7 (5). Public Library of Science: e34861. doi:10.1371/journal.pone.0034861.
Pachkov, Mikhail, Ionas Erb, Nacho Molina, and Erik van Nimwegen. 2007. “SwissRegulon: A Database of Genome-Wide Annotations of Regulatory Sites.” Nucleic Acids Research 35 (suppl 1): D127–31. doi:10.1093/nar/gkl857.
Pagès, Hervé. 2014. SNPlocs.Hsapiens.dbSNP142.GRCh37: SNP Locations for Homo Sapiens (DbSNP Build 142).
Richter, Klaus, Martin Haslbeck, and Johannes Buchner. 2010. “The Heat Shock Response: Life on the Verge of Death.” Molecular Cell 40 (2). Elsevier: 253–66. doi:10.1016/j.molcel.2010.10.006.
Ritchie, Matthew E, Belinda Phipson, Di Wu, Yifang Hu, Charity W Law, Wei Shi, and Gordon K Smyth. 2015. “limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies.” Nucleic Acids Research 43: doi: 10.1093/nar/gkv007.
Ritossa, Ferruccio. 1962. “A New Puffing Pattern Induced by Temperature Shock and DNP in Drosophila.” Experientia 18: 571–73.
Sfar, Sana, Hamadi Saad, Faouzi Mosbah, and Lotfi Chouchane. 2010. “Synergistic Effect and VEGF/HSP70-Hom Haplotype Analysis: Relationship to Prostate Cancer Risk and Clinical Outcome.” Human Immunology 71 (4): 377–82. doi:http://dx.doi.org/10.1016/j.humimm.2010.01.017.
Stojnic, Robert, and Diego Diez. 2014. PWMEnrich: PWM Enrichment Analysis.
Vihervaara, Anniina, Christian Sergelius, Jenni Vasara, Malin A. H. Blom, Alexandra N. Elsing, Pia Roos-Mattjus, and Lea Sistonen. 2013. “Transcriptional Response to Stress in the Dynamic Chromatin Environment of Cycling and Mitotic Cells.” Proceedings of the National Academy of Sciences 110 (36): E3388–97. doi:10.1073/pnas.1305275110.
Styled with knitrBootstrap