Introduction to eQTL analysis
Peter Humburg
Overview
- Hands-on : Setup
- What are eQTL?
- Detecting eQTL – A simple model
- Hands-on : Simple linear regression
- Detecting eQTL – Not that simple
- Hands-on : Covariates
- Covariates – Choose wisely
- If only we knew – Covariates for real data
- Hands-on : Dealing with real data
- Large scale eQTL analysis
- Hands-on : Scaling it up
- Interpreting results
- Summary
Expression quantitative trait loci
If the trait of interest is the expression of a gene, we talk about eQTL.
- Associations can be local (cis)
- or distant (trans)

Expression quantitative trait loci
If the trait of interest is the expression of a gene, we talk about eQTL.
- Associations can be local (cis)
- or distant (trans)
Expression quantitative trait loci
If the trait of interest is the expression of a gene, we talk about eQTL.
- Associations can be local (cis)
- or distant (trans)
How do we know whether a locus is associated with the expression of a gene?
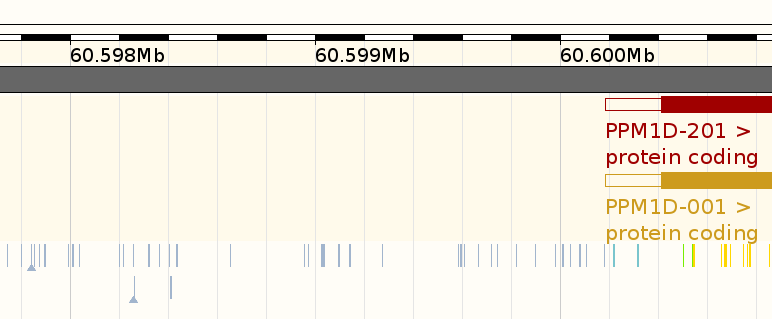
- Any given locus contains multiple SNPs.
- Can determine genotypes for these SNPs in a (large) number of individuals.
- Measure gene expression for genes of interest.
- Assess the evidence that expression varies with genotype.
How do we know whether a locus is associated with the expression of a gene?
- Any given locus contains multiple SNPs.
- Can determine genotypes for these SNPs in a (large) number of individuals.
- Measure gene expression for genes of interest.
- Assess the evidence that expression varies with genotype.
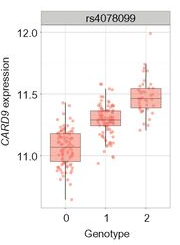
Linear additive model
Different alleles of a SNP may exhibit a dosage effect.
- Using the AA genotype as baseline, each copy of the B allele changes expression by a fixed amount.
- Implies a linear relationship between the mean gene expression and the number of B alleles.
- Estimating the change in expression due to the B allele to quantify the SNP’s contribution to gene expression.
Linear regression – Robustness
Linearity
If the true relationship between \(Y\) and \(X\) is non-linear conclusions may be misleading.
When might this occur with eQTL data?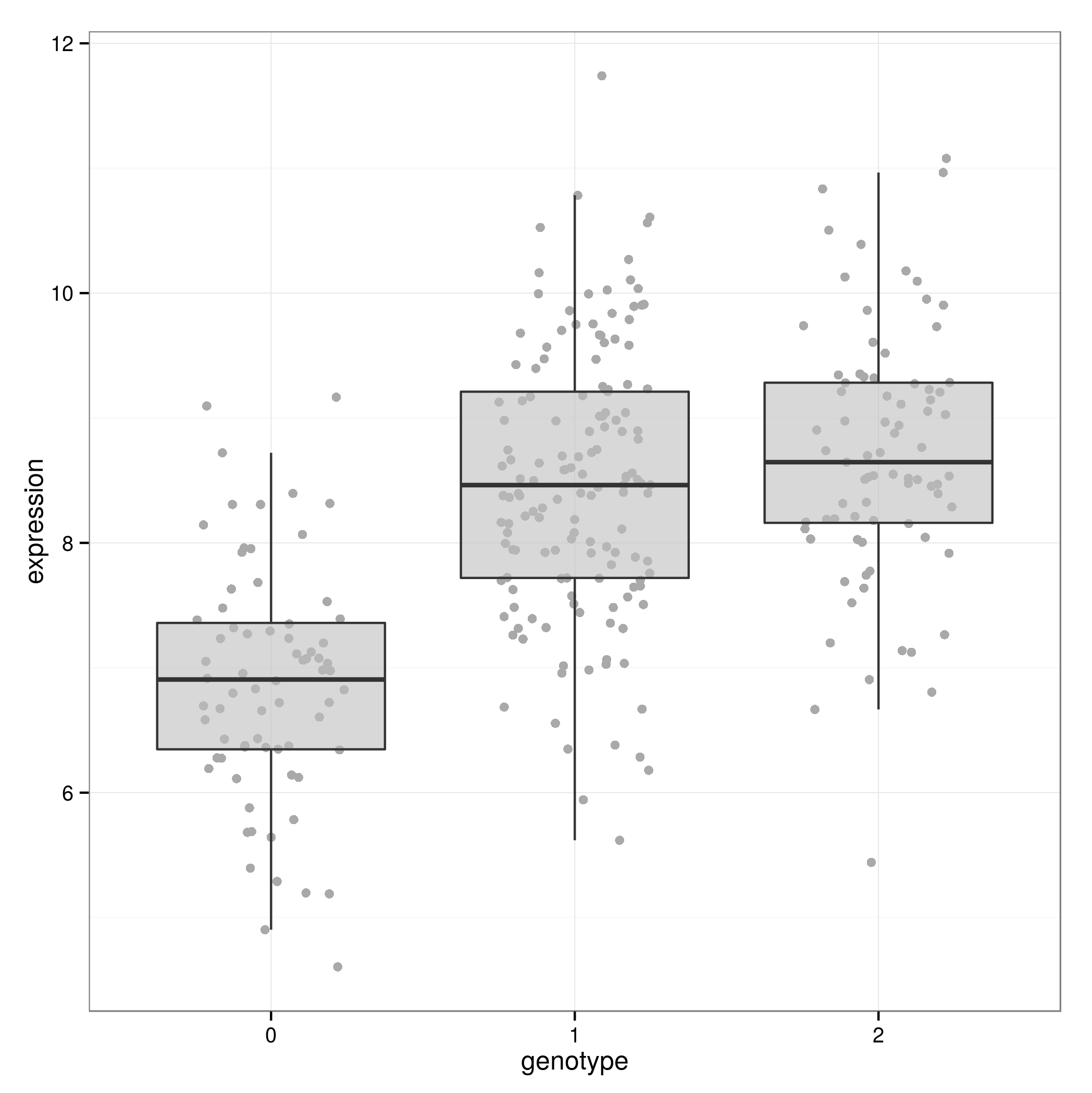
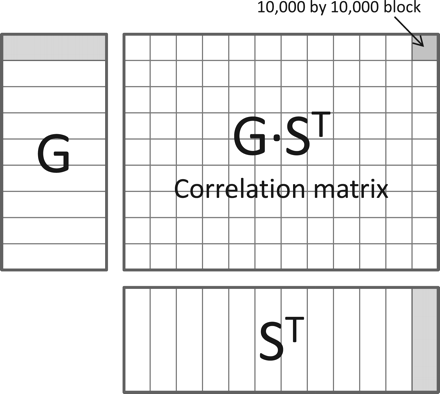
Fast model fitting with Matrix-eQTL
Matrix-eQTL strategies to reduce run time:- Required test statistic can be expressed in terms of the correlation between SNP and gene expression.
- Use efficient matrix multiplications to compute the correlations.
- Computing p-values is expensive, only do this for SNP/gene pairs that are sufficiently interesting.